
将治疗和成像分子组装成无机纳米载体的策略
1.介绍
化疗是癌症最常见的治疗模式,不仅对转移病灶有效,而且会对快速生长的健康组织造成不良影响。研究人员致力于改善癌症化学治疗的局限性,包括半衰期短、组织分布广和缺乏靶向性,这些都是治疗(抗癌)活性弱和严重不良反应的原因。此外,这些缺点需要服用大量药物来达到所需的治疗效果,这并不划算,而且往往会对健康组织产生不希望的毒性作用。化疗药物的剂量限制溶解度是一个挑战。口服和静脉给药是两种最流行的方法。然而,这些方法确实有一些缺点。例如, 由于这些药物与体内代谢途径的相互作用,口服这些药物会破坏药代动力学。因此,可能会使用比必要更高的药理学剂量,这可能会导致更大的毒性。通常,疏水性药物的配方需要使用溶剂(由于其溶解度差),这会加剧其毒性作用。为了使药物在血液中达到最佳浓度并产生足够的药理作用,药物的溶解度至关重要。例如,口服水溶性较差的药物需要大量剂量才能达到治疗性血浆水平。药物的发现和开发受到其低水溶性的影响。任何需要被身体吸收的药物都必须是水性的,以确保完全吸收。传统的静脉注射(i.v.)路线有其自身的困难。一些传统的静脉注射药物的特异性较低。这种非特异性药物分布限制了癌症细胞内的治疗效果,同时对健康细胞、组织和器官造成相当大的毒性,导致各种负面副作用,包括脱发、虚弱、恶心、呕吐和器官功能障碍,所有这些都导致癌症患者的生活质量低下。此外,癌症细胞膜中过表达的多药耐药相关蛋白(MRP)或P-糖蛋白(P-gp)为从细胞中去除化疗药物提供了途径,从而诱导多药耐药(MDR),并使胞浆中的药物不足以达到所需的治疗效果。与化疗药物相关的这些局限性导致人们对使用纳米粒子(NP)作为各种治疗药物的载体越来越感兴趣,包括小分子抗癌药物、siRNA、miRNA、mRNA、质粒和蛋白质。纳米载体由于其延长的血液循环半衰期和改善的生物相容性,以及改善的脱靶分布和在肿瘤中的更高积聚,提供了增强的治疗效果,从而显著减少了药物的不良反应。虽然过小的纳米载体(即-5nm)会被肾脏排泄,但较大的纳米载体(250nm-3µm)通常会被单核吞噬系统(MPS)识别,从而激活关键的防御机制并将NP从体内清除。脾脏、肝脏和淋巴结中的MPS倾向于清除和降解血液中的异物。
无机NP引起了人们的广泛关注,因为它们的大表面积与体积比能够有效地装载治疗药物。此外,无机NP的其他独特特性,包括其易于表面改性、惰性、可调尺寸和形态、高稳定性、多孔性以及减轻化疗相关副作用和药物降解的能力,使其成为治疗恶性细胞的有前景的候选药物。显然采用其中一些特征的无机纳米载体正在进行临床试验,而其他载体则正在广泛研究其潜在的临床应用。然而,在批准用于临床研究的纳米载体中,由于癌症细胞的异质性未被充分探索,只有少数显示出积极的结果,这改变了NP的摄取和定位,从而损害了其治疗效果。此外,由于对动物和人类研究中的生理和病理变化了解不足,有必要对患者进行仔细筛查,以确定哪些患者最有可能对纳米疗法产生反应。从这个意义上说,这些治疗方法与已批准或正在开发的基于生物标志物治疗特定患者群体的疗法相当。因此,恶性肿瘤的生物学基础以及患者内部都存在很大的复杂性,这会影响NP的分布和疗效。因此,动物模型和人类研究之间的翻译差距给靶向治疗中的药物递送系统带来了严重的问题。
无机NP作为药物载体比有机纳米载体具有优势。下面列出了无机NP的一些优点:
•表面改性:应用于无机NP的大多数改性都能保护生物分子免受酶降解。即使是涉及氯化钠的修改也是如此。例如,发现层状双氢氧化物(LDH)NP在微酸性室(pH=4.5-6.0)中逐渐溶解。LDH-NP的分解可能会提高pH值,导致随后的溶解速度减慢。这允许DNA和其他生物分子受控释放到细胞中。无机纳米载体的表面改性选项比有机纳米载体的更为多样化。由于易于表面官能化和独特的物理化学特性,已经探索了多种将治疗剂与无机NP结合的方法。为了实现响应性释放,治疗剂通常通过与特定官能团的疏水和共价相互作用与无机NP结合,这些官能团可以被酶或其他刺激物切割。
•稳定性:大多数无机NP表现出化学稳定性,LDHs除外。这是一个很好的特性,因为它确保了它们的物理化学性质在将治疗药物输送到目标部位的整个过程中 保持不变。只要NP具有高化学稳定性,它们就不会在人血浆和细胞质中降解。因此,这些NP要么在细胞中积聚,在血液中循环,要么被代谢。通过用聚乙二醇(PEG)等聚合物修饰无机NP的表面,可以提高其在血液中的稳定性和延长循环时间。此外,抗体、配体和其他分子也可以附着在无机NP表面进行主动靶向。基于以下因素,无机纳米载体的靶向递送可以最大限度地提高其治疗效果延长血液循环时间和主动靶向纳米载体。此外,许多方法已被应用于无机纳米载体,以释放由外部刺激引发的药物。相比之下,有机NP,如聚合物胶束,在静脉注射胶束溶液后,由于血液的严重稀释,容易变形和分解,从而可能导致负载药物的泄漏和随后的排放。目前,这种限制可以通过胶束外壳的化学共轭或交联来改善药物-聚合物相互作用。此外,有机NP的稳定性在高温下会降低。这表明无机纳米粒子是治疗癌症的更好选择。
•毒性:无机NP的生物相容性和低细胞毒性使其成为优秀的递送载体。生物相容性无机NP包括Au(1-100nm)、碳纳米管(CNT;1-100纳米)、LDH(30-200nm)和Fe3O4(1-50nm)。与阳离子有机载体(10µg·mL-1)相比,它们的LD50(或LD80)在约1mg·mL-1的浓度下非常高,而阳离子有机载体的LD50或LD80浓度约为10µg•mL-1。研究人员发现,乳铁蛋白、铜蓝蛋白、普鲁兰、右旋糖酐和白蛋白修饰的磁铁矿NP强烈粘附在人成纤维细胞上,阻止其内化到细胞中,从而增强细胞活力。因此,对无机NP进行适当的修饰可以将细胞活力恢复到正常水平。大多数有机聚合物NP由聚苯乙烯(PS)、聚甲基丙烯酸甲酯(PMMA)和聚丙烯酰胺(PAA)等聚合物组成,这些聚合物是不可生物降解的。由于难以从体循环中排泄这些聚合物NP,它们往往会在体内积聚并引起毒性。脂质体可以根据其组成、平均直径或表面电位诱导毒性。例如,阳离子脂质体可以与血清蛋白、脂蛋白和细胞外基质(ECM)相互作用,导致聚集形成并在到达靶点之前释放药物,从而由于它们与体内不同组织的脱靶相互作用而引起严重的不良毒性。一项研究表明,包封在聚乙二醇化脂质体中的唑来膦酸(ZOL)在体内引起了不希望的毒性。
•光学和磁性:一些无机纳米载体具有成像、磁性和光学特性,是癌症治疗的理想选择。它是报道了纯抗体偶联的氧化铁NP(10nm核心尺寸)靶向多形性胶质母细胞瘤(GBM)细胞中的表皮生长因子受体缺失突变体vIII(EGFRvIII)进行靶向治疗,并增强实验性胶质母瘤的磁共振成像(MRI)对比度。用纳米载体治疗后,胶质母细胞瘤细胞存活率显著降低。此外,人星形胶质细胞在NP偶联物治疗下没有毒性。发现磁NP的MRI引导对流增强递送(CED)可以提高致瘤性胶质母细胞瘤异种移植物(U87-DeltaEGFRvIII)的存活率。使用与金纳米颗粒结合的抗EGFR抗体,进行了另一项研究,以探索金纳米颗粒用于生物成像的光学和电子特性。在最佳表面等离子体散射条件下,使用传统显微镜在白光照射下获得图像,从而在深色背景上产生AuNP的彩色图像。此外,无机NP比有机NP具有更高的量子产率、更长的保质期和优异的光稳定性。
除了复杂的生物系统外,转移性肿瘤中所需治疗量的化疗药物的积累取决于它们与载体的相互作用,在到达靶点之前影响生物系统中合成复合物的稳定性,以及随后治疗有效载荷的释放。因此,了解将治疗剂和成像分子装载或封装到无机纳米载体中的不同方法至关重要,以最大限度地提高药物装载效率,延长药物的血浆半衰期,并增强其在靶点的分布和递送。药物负载到NP中可以改变复合物的大小、形态和表面性质,从而改变其药代动力学,对血浆清除和肿瘤靶向产生影响。此外,靶位点纳米载体的药物释放可能取决于将药物组装成颗粒的各种策略。需要解决这些新出现的主题,以实现无机纳米载体在癌症精准治疗中的临床转化的里程碑。本综述重点介绍了用于不同无机纳米颗粒的各种负载机制,特别是涉及物理包埋到多孔/中空纳米结构中、与天然和表面改性纳米颗粒的离子相互作用、与表面功能化纳米材料的共价键合、疏水键合、基于亲和力的相互作用,以及通过共沉淀或阴离子交换反应进行插层。
2.将治疗和成像分子装载到无机NP中的原理
采用各种策略和相互作用将治疗剂和成像剂装载在不同类型的无机NP中,通过增加其溶解度、稳定性和血液循环时间(血浆半衰期)来调节装载分子的药代动力学特征。交互的主要类别包括:
•小分子药物的包封或包封
•亲和互动
•通过共沉淀或阴离子交换反应进行插层
•共价债券联系
•离子相互作用
图1突出了将药物装载到无机NP中所采取的不同策略。
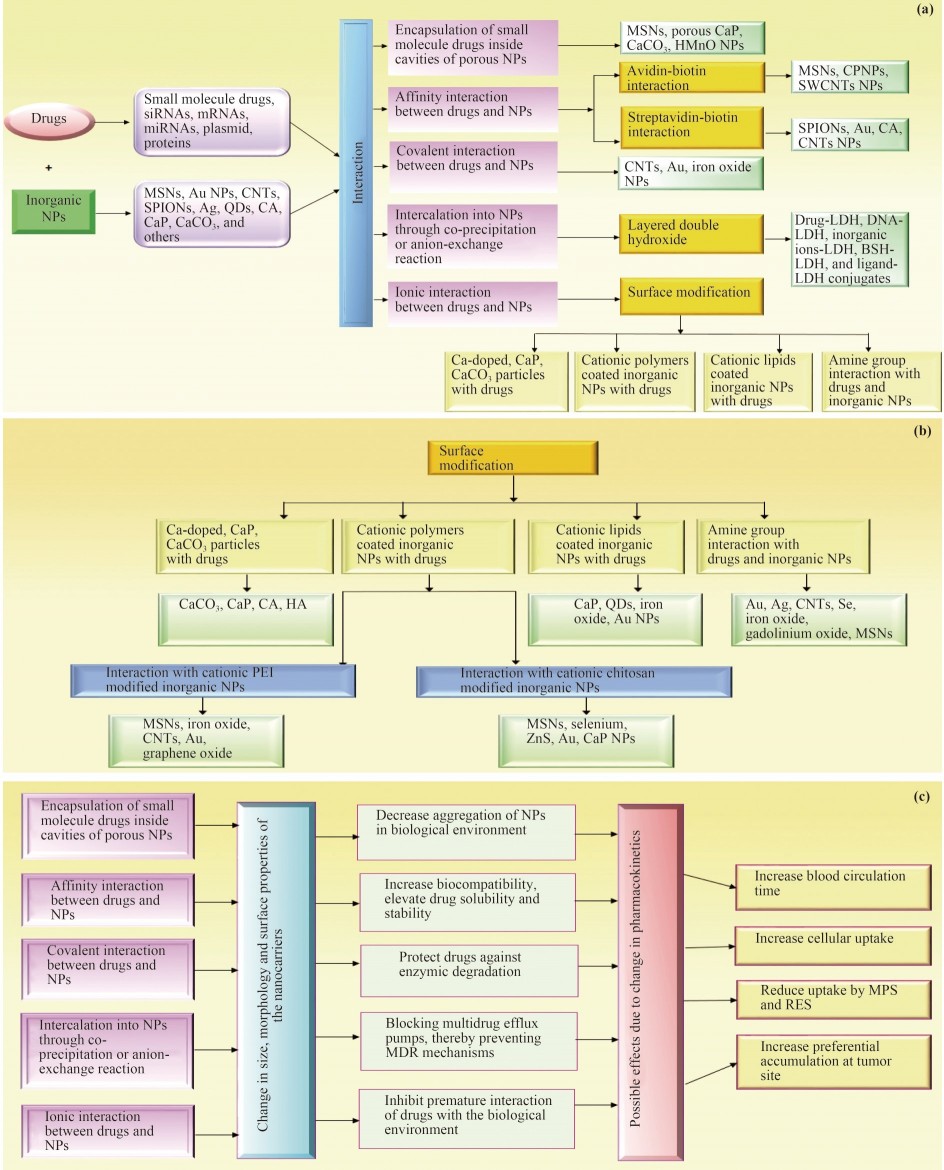
图1(a)将治疗剂和成像分子组装成无机NP的不同策略的示意图。
(b)无机纳米颗粒和药物之间的不同表面改性方法。
(c)由于无机纳米载体与药物的相互作用(通过不同的方法),其尺寸、
形态和表面性质发生了变化,突显了它们对药代动力学的可能影响。
注:银;Au,金;CA,碳酸盐磷灰石;CaCO3、碳酸钙;CaP,磷酸钙;CNT、碳纳米管;
CPNP,磷酸硅酸钙纳米粒子;羟基磷灰石;HMnO,空心氧化锰;LDH,层状双氢氧化物;MDR,多药耐药性;
MPS,单核吞噬系统;MSN,介孔二氧化硅纳米粒子;QD,量子点;RES,网状内皮系统;
SPION,超顺磁性氧化铁纳米粒子;SWCNT,单壁碳纳米管;ZnS、硫化锌。
3.多孔纳米材料腔内小分子药物的容纳
设计化学稳定的纳米载体,在包封药物运输到靶位点的过程中保护它们,这一点至关重要。NP药物相互作用的强度和性质也会影响药物的释放速度。此外,刺激反应性连接体或表面功能化的孔,将药物附着在其核心,可用于改变释放速率。孔工程有助于生成能够用特定化学结构包裹药物的特定多孔结构。大多数情况下,介孔二氧化硅纳米粒子(MSNs)、多孔磷酸钙(CaP)和碳酸钙( CaCO3)、中空氧化锰(HMnO)和中空金纳米粒子(HGNP)作为药物包封的纳米载体系统越来越受到人们的关注。重要的是,对于多孔纳米材料来说,腔结构的设计和合成对其功能化至关重要,因为它们有助于调节原子-分子和分子-分子之间的相互作用、原子和分子之间的热/统计力,并协调腔单元内和腔单元之间的原子或分子的作用。
3.1
MSNs包封核酸和药物的能力是由于其独特的多孔结构、大的内外表面积、增加的孔体积、可调的孔径、灵活的表面改性、可忽略的毒性、优异的生物相容性和窄的尺寸分布。MSNs的选择性表面功能化使它们能够实现特定的药物相互作用,这反过来又使它们能够控制药物的释放。在大多数情况下,这些是决定有资格通过MSN交付的药物类型和数量的主要因素。为了防止过早释放,必须确定治疗药物的孔径范围。相反,中尺度通道可以防止药物结晶,从而增强其释放。理想情况下,孔径和分子药物尺寸之间的比值必须大于1,才能获得最佳的药物负载量。由于扩散增加,药物的负载率随着该比率的增加而增加。通过调节表面活性剂的浓度可以改变孔的大小。MSN的孔径受到实验参数的影响,如催化剂浓度、二氧化硅前体和反应温度/时间。此外,不同的孔几何形状,无论是二维(2D)还是三维(3D)结构,对药物负载和释放都有显著影响。中孔的大表面积会影响颗粒与其周围环境在体内的相互作用。此外,MSNs可以加速肾上腺素的氧化,从而在孔隙表面产生活性氧(ROS)。因此,孔隙率增加的中孔结构毒性较小。重要的是,其他颗粒制剂尚未通过表面负载、基质负载和空腔负载机制实现药物在MSN中的包封。有效载荷从MSN的释放可以通过一个仅在外部刺激下打开的帽结构来启动,并且可以使用“看门人”策略或修改孔的内表面来控制药物的结合亲和力来进行调节(图2)。图2描绘了各种MSN药物递送方法的图示。
在靶向MSNs药物递送中,药物过早释放是一个重大挑战。最典型的药物装载方法是将药物吸附到孔中,然后药物溶解,在所需位置释放货物。在这方面,有几个
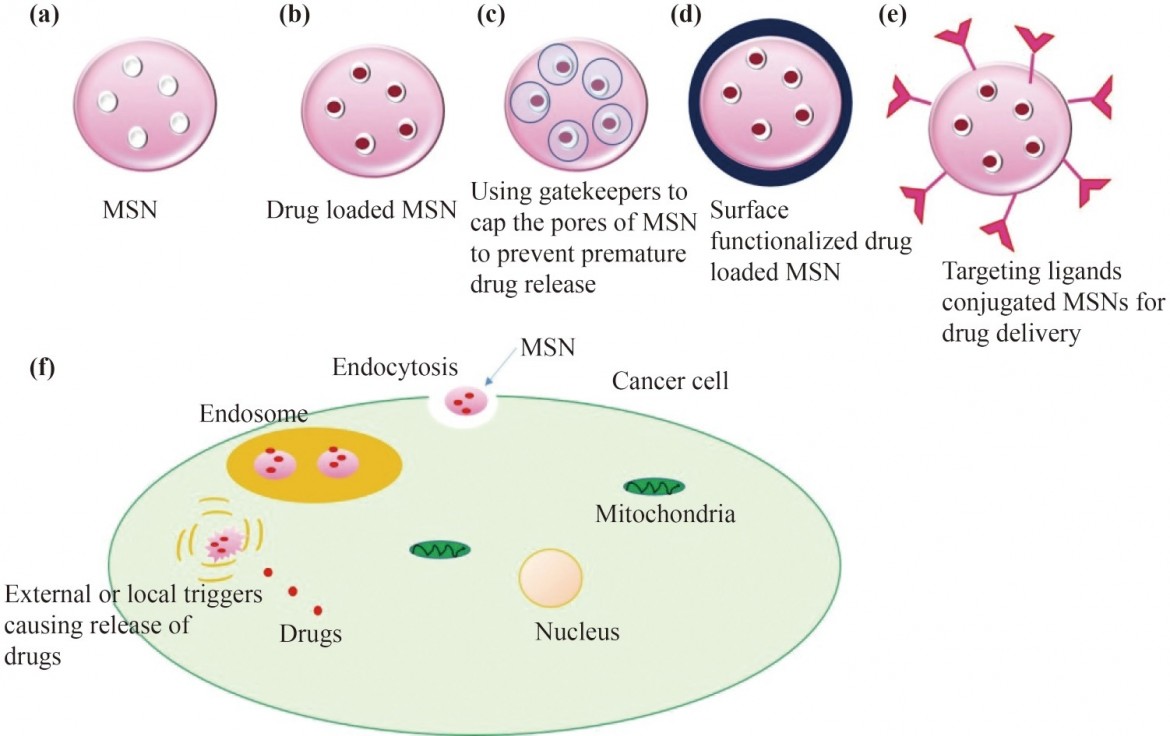
图2示意图:(a)MSN;(b)载药MSN;
(c)使用看门人来盖住MSN上的毛孔,以防止药物过早释放;
(d)负载药物的MSNs已被表面功能化,以减少脱靶相互作用并增强位点特异性特征;
(e)配体偶联的MSNs可以通过内吞作用促进有效的细胞内化,以便在靶位点递送药物;
(f)MSN通过内吞作用进入癌症细胞,并在外部或局部刺激下释放药物。
MSN药物递送系统(DDSs)的设计具有孔隙守门人。聚合物超分子结构、DNA结构和蛋白质是看门人的例子。各种刺激,包括酶、光照射、磁场、pH值、氧化还原化学和竞争性结合,都会引发这些药物的释放。MSN大多涂有聚乙烯亚胺(PEI),以覆盖孔隙的开口,通过“分子门控”增强货物滞留,并通过内吞促进NP的细胞摄取(图3)。有趣的是,一个研究小组发现,通过非共价连接用PEI使MSN表面功能化显著增加了细胞摄取,并形成了一个带正电的DNA或小干扰RNA(siRNA)结合体。观察到用10kDPEI包覆通过用siRNA转导HEPA-1细胞有效地降低了绿色荧光蛋白(GFP)的表达。另一组将PEI/PEG/MSN与曲妥珠单抗偶联,以有效运输siRNA。NP的流体动力学直径为~100nm(具有三个均匀大小的MSN核心材料)和200nm(MSN的非均匀大小核心材料)。重要的是,由于其阳离子性质,配方中的PEI有助于产生正表面电荷。PEI/PEG/MSNs/siRNA构建体在体外成功诱导HER2阳性细胞凋亡。因此,多次静脉注射PEI/PEG/MSNs/siRNA构建体抑制了原位HCC1954荷瘤小鼠的肿瘤发展三周。当与来自外周血的人类单核细胞接触时,PEI/PEG/MSNs/siRNA构建体具有较低的细胞因子诱导和生物相容性。因此,PEI-包被的MSNs可以增加细胞摄取,这反过来可能导致癌症细胞凋亡。王及其同事通过可裂解的二硫键将MSN的出口与聚丙烯酸(PAA)接枝。PAA被选为防止药物进入MSN中孔的看门人,主要是因为它具有许多优点,包括阻断MSN进入点的适当分子量、高生物相容性,以及延长血液循环周期和使MSN在生理条件下更稳定的能力。在这项研究中,荧光染料RhB被用作模型药物。基于体外释放动力学,发现RhB在没有谷胱甘肽(GSH)的中孔或生理pH(7.4)的磷酸盐缓冲盐水(PBS)中受到显著限制。相比之下,当补充GSH或pH5.0的PBS时,RhB的释放显著增加。此外,在GSH和PBS(pH5.0)同时存在的情况下
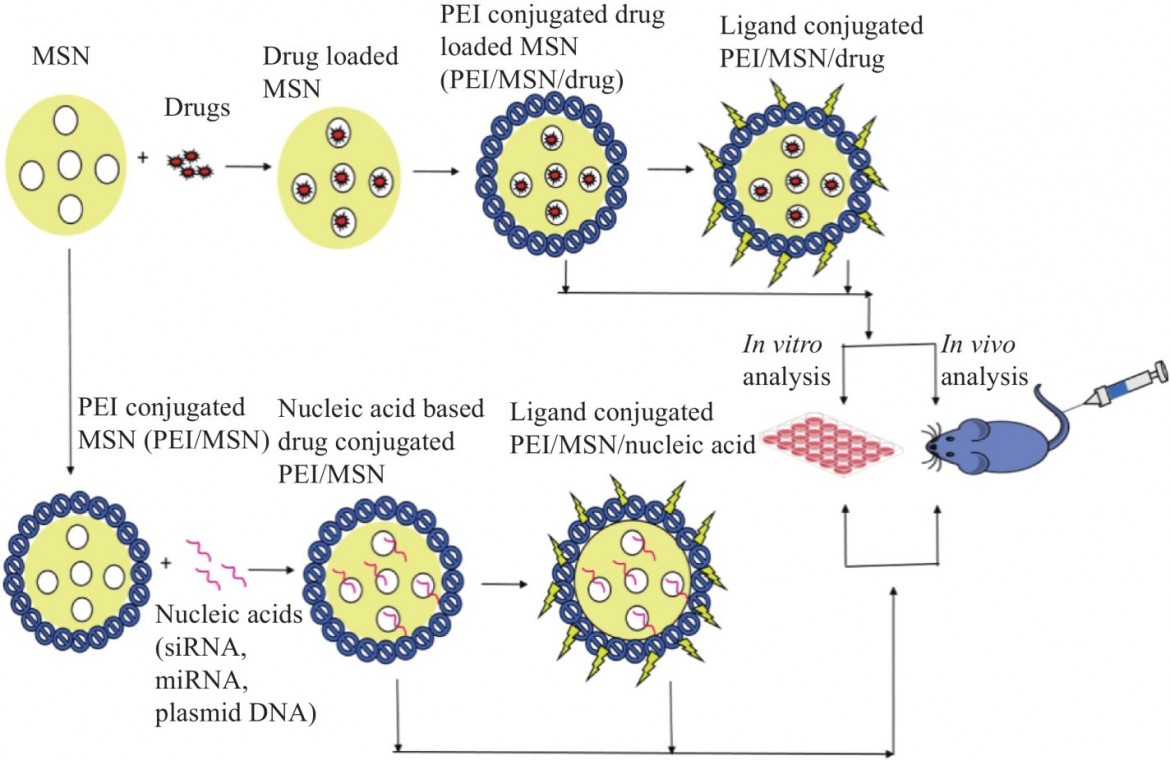
图3用于体外和体内分析的装载货物并经配体和无配体修饰的PEI偶联MSN的示意图。
RhB的释放进一步增强。这些发现表明,这种药物递送方法具有双重反应药物释放特性,可能是癌症治疗的好选择。
据报道,阳离子聚合物涂层可以提高MSNs的转染效率和DNA负载量。在一项研究中,MSN上的聚酰胺胺(PAMAM)树枝状大分子涂层(平均尺寸250nm,平均孔径2.7nm)允许DNA络合的阳离子结合。这种方法确保了装载DNA的开放孔。在HeLa和CHO细胞中,共聚焦荧光显微镜(CFM)和透射 电子显微镜(TEM)证实了树枝状大分子(G2)-MSN-DNA复合物的有效内吞作用。生物相容性研究证实,有和没有G2MSNs的HeLa培养物中的增殖是相似的,表明G2MSNs不会干扰细胞生长。因此,MSNs在体外没有毒性,可以作为防止酶切割的DNA保护剂。在另一项研究中,如Kar等人所报道的,将聚-L-精氨酸(n=10和20)接枝到MSN的表面,以成功递送药物或质粒DNA。用阳离子官能团对MSNs进行表面修饰可以与带负电荷的核酸结合,促进细胞膜的快速渗透。MSN-p(LArg)20和MSN-p的ζ电位分别为(+32±4)mV和(+20±2)mV。这两种颗粒都是离散的,MSN-p(LArg)20的粒径分别为17nm(TEM和扫描电子显微镜(SEM))和80nm(动态光散射(DLS)。此外,MSN-p(LArg)颗粒用于在细胞内递送mCherryDNA质粒,导致mCherry蛋白表达。在HeLa和A549细胞中,MSN-p(LArg)在100µg·mL-1MSN浓度下显示出更高(85%)的细胞存活率和更高的细胞摄取率(大于90%)。使用阿霉素(DOX)的初步数据显示,DOX-MSN-p(LArg)可以有效提高治疗效果,并导致HeLa和A549细胞凋亡。MSN毛孔中DOX含量增加,可以显著提高治疗效果。因此,孔径更大的MSN可能会产生更有希望的效果。核酸和药物的结果表明,MSN偶联物可以成为向癌症细胞递送基因和药物的重要载体。
为了开发一种pH依赖性的细胞内载体,用于靶向肿瘤细胞核内DOX的分布,Zou等人用明胶封端MSN(MSN@Gelatin)厚度约为8nm。表面电荷MSN@Gelatin被发现是电负性的(-0.226mV)。在生理pH下,由于明胶涂层,包封的DOX没有释放,而在酸性pH下,静电排斥明胶和MSN之间的涂层被打开,释放了包埋在其中的DOXMSN@Gelatin系统。通过紫外-可见吸收测量,该系统显示DOX包封率高达47.3mmol·g−1SiO2。通过内吞作用进行的细胞内化揭示了DOX/MSN@Gelatin在溶酶体中的成功积累,随后酸诱导DOX摄取到Hep-G2肝癌细胞的细胞核中。对于DOX/MSN@Gelatin,记录了剂量依赖性细胞毒性(IC50=(17.27±0.63)µg·mL-1)。相反,在以下情况下观察到最小毒性(IC50>100µg·mL-1)MSN@Gelatin.在另一项研究中,甘露糖基化聚乙烯亚胺(MP)与MSN偶联,产生MPS偶联物,用于体外质粒DNA转染,靶向含有甘露糖受体的巨噬细胞。细胞毒性研究显示,与PEI25K(载体)相比,MPS/DNA复合物的活细胞百分比很高。MPS/DNA复合物转染HeLa和Raw264.7细胞系的有效性通过受体介导的内吞作用得到增强。当在MCF-7/MDR荷瘤异种移植物模型中检测Dox和P-gpsiRNA负载的PEI-PEG-MSNs复合物时, 由于血液循环延长,观察到肿瘤中的积聚增强,与游离Dox(17%)、MSN-Dox(62%)和P-gpsiRNAs(0%)相比,肿瘤生长抑制率为80%。
同样,脂质体包裹的介孔二氧化硅核壳磁性NP,Lipo(负ζ电位)用于偶联和递送曲妥珠单抗(Lipo-Her2Ab)用于体外靶向Her2/neu-过表达的乳腺癌症细胞。MRI和CFM均用于评估Lipo的靶向性-Her2Ab。在Her2/新阳性癌症细胞中,Lipo-Her2Ab被发现显著积累,这表明它们在体外具有药物递送和多模式成像的潜力。Sun等人的研究表明,钆(Gd)掺杂的MSNs负载了DOX,并与含有吲哚青绿(ICG)的热敏脂质体(TSL)结合(DOX@GdMSNs-ICG-TCLs)可以用作三模态成像引导的纳米平台。它们的平均粒径为233.8nm,表面电荷为-25.2mV。为了防止DOX泄漏并提高细胞摄取,DOX@GdMSNs用含有叶酸(FA)的TSL包覆。在近红外(NIR)照射下,ICG产生热量并破裂ICGTSL,有效释放DOX。不用说,由于MSN的成像能力,其位点特异性靶向的潜力可能会提高癌症的治疗效果。
3.2 CaCO3和CaP
值得注意的是,其他通常研究的纳米多孔载体包括CaCO3和CaP,它们经常用于基因递送。然而,CaCO3-NP比CaP-NP更频繁地被利用,因为它们具有更多孔的结构、更大的表面积、生物相容性、pH敏感性以及负载和释放药物的能力。此外,制备CaCO3/DNA共沉淀物不需要添加缓冲溶液来调节pH值,而合成CaP/DNA共沉淀物则需要。例如,羧甲基壳聚糖(CMC)改性的CaCO3微/纳米球分别通过毛细管力和CMC和DOX的正负电荷之间的静电吸引来包封DOX。由于CMC和DOX之间的静电相互作用以及多孔结构的存在,DOX的包封效率大大提高了60%以上。此外,研究人员还专注于使用CaPNP作为成像平台和药物载体的创新方法。例如,一项研究强调了CaP与有机分子(约27nm)的包封,由于其生物相容性和pH溶解特性,可以在细胞内分布和成像,从而在肿瘤内以有规律的方式释放治疗性物质。
3.3 HMnO和HGNP
另一个感兴趣的领域包括通过阳离子PEI-3,4-二羟基-L-苯丙氨酸(DOPA)偶联物对无机NP进行表面改性,用于络合治疗。为了促进肿瘤靶向,Bae等人通过MRI研究了HMnONP在癌症治疗和成像中的有效性。首先,使用DOPA作为粘合剂部分制备带正电的HMnOs。接下来,通过傅里叶变换红外光谱(FTIR)和X射线光电子能谱(XPS)证实,利用DOPA的重要金属氧化物结合亲和力,将阳离子PEI-DOPA偶联物固定在HMnO-NP表面上。为了将靶向siRNA转运到乳腺和卵巢癌症细胞中,使用马来酰亚胺-PEG-琥珀酰亚胺基碳酸酯(SC)将NP与赫赛汀在表面偶联。共聚焦显微镜MRI显示,结合赫赛汀可以增强SK-BR3细胞(HER2+)对修饰HMnOs的摄取。与MCF-7(HER2−)细胞相比,这反过来在使用T1加权MRI检测癌症细胞方面发挥了至关重要的作用,表明赫赛汀功能化NP显示出靶向性。类似地,一项研究证明了DOX负载的PEG化多功能HGNP(DOX-HGNP)的发展,其在NIR照射时释放DOX并产生热量,导致DOX和高温诱导的对患有肺癌的A549异种移植物小鼠的有害影响。
4.亲和互动
最近,在开发合适的药物载体方面,无机NP中负载药物的亲和相互作用(抗生物素蛋白/生物素或生物素/链霉抗生物素)经历了复兴。Avidin是一种存在于鸟类、爬行动物和两栖动物卵中的基本四聚体糖蛋白。生物素,通常被称为维生素H、维生素B7或辅酶R,具有一种环结构,即四氢噻吩与四氢咪唑酮结合。生物素部分可能被证明在癌症治疗中有用 ,因为它在增殖的癌症细胞中比在正常细胞中更频繁地表达。抗生物素蛋白的高正电荷改善了生物素包裹的NP在细胞中的内化。相比之下,与生物素化细胞系进行生物偶联的抗生物素蛋白孵育可实现近100%的表面粘附和内吞作用。链霉抗生物素蛋白也被证明在体内特定组织中积累,特别是在恶性肿瘤中。因此,一些基因治疗载体可以单独使用抗生物素蛋白靶向特定组织,同时使用蛋白质的生化变化改变组织特异性。抗生物素蛋白/生物素相互作用在治疗作用中具有巨大的前景,因为它是一种通用、特异和稳定的高亲和力相互作用,对变性化学物质、pH值、蛋白水解酶、温度和刺激性有机试剂具有抗性。重要的是,成像剂和靶向配体通常用于抗生物素蛋白NP平台,将药物递送到特定部位。这些基于抗生物素蛋白的平台对正常组织的毒性较低,但对恶性肿瘤的毒性较高。体内配体或组织的生物素化可以利用强烈的抗生物素蛋白/生物素相互作用进行靶向递送。值得注意的是,链霉亲和素是抗生物素蛋白最常见的类似物。链霉抗生物素蛋白(源自阿维迪尼链霉菌)是一种56kDa的非糖基化四聚体蛋白,等电点(pI)值约为5-6。应该指出的是,抗生物素蛋白/生物素和生物素/链霉抗生物素系统表现出很强的结合力和特异性,使其能够用生物素化试剂和生物分子如DNA低聚物、肽、荧光染料和抗体进行修饰。 图4显示了抗生物素蛋白/生物素和链霉抗生物素素/生物素复合物的形成,并描述了它们之间的一般相互作用。
4.1 超顺磁性氧化铁纳米粒子(SPION)
SPION是众所周知的MRI材料,以其在T2(自旋)时的出色弛豫性能而闻名。因此,它们高度疏水表面的聚集、巨噬细胞的吸收和网状内皮系统(RES)的消除限制了它们的利用。为了解决这些局限性,开发了许多基于亲和相互作用的SPION药物递送平台。一项早期研究揭示了放射性活性纳米探针的形成,以识别临床前肿瘤模型中的乳腺癌症细胞,通过生物素-链亲和素相互作用将soiTrastuzumab加载到SPION中,以增强细胞标记。此外,该方法成功检测到HER2受体。然而,这些利用生物素-链霉抗生物素蛋白相互作用的探针可能会在人类中引发深刻的免疫反应。有趣的是,这种方法中使用的超顺磁性氧化铁颗粒的分子直径约为50nm(分子量:40-50MD),因为更强的弛豫性和减小的分子尺寸可以促进有效的药物递送。
生物标志物可以在MRI中用NP预靶向,以帮助体内软组织的精确诊断方法,提供具有出色空间分辨率的高对比度成像。刘的研究小组用1,2-二硬脂酰-sn-甘油-3-磷酸rac-甘油钠盐(DSPG)、胆固醇和1,2-二硬脂酰基-sn-甘油3-膦酸-N-醇胺-N-(生物素-PEG2000-DSPE)将SPION修饰成磁性脂质体。生物素部分的表面功能化使得通过预靶向研究肿瘤中的血管生成成为可能。脂质体双层和PEG修饰阻止了巨噬细胞摄取Fe3O4核。细胞毒性试验表明,所制备的磁性脂质体具有超顺磁性和生物相容性。
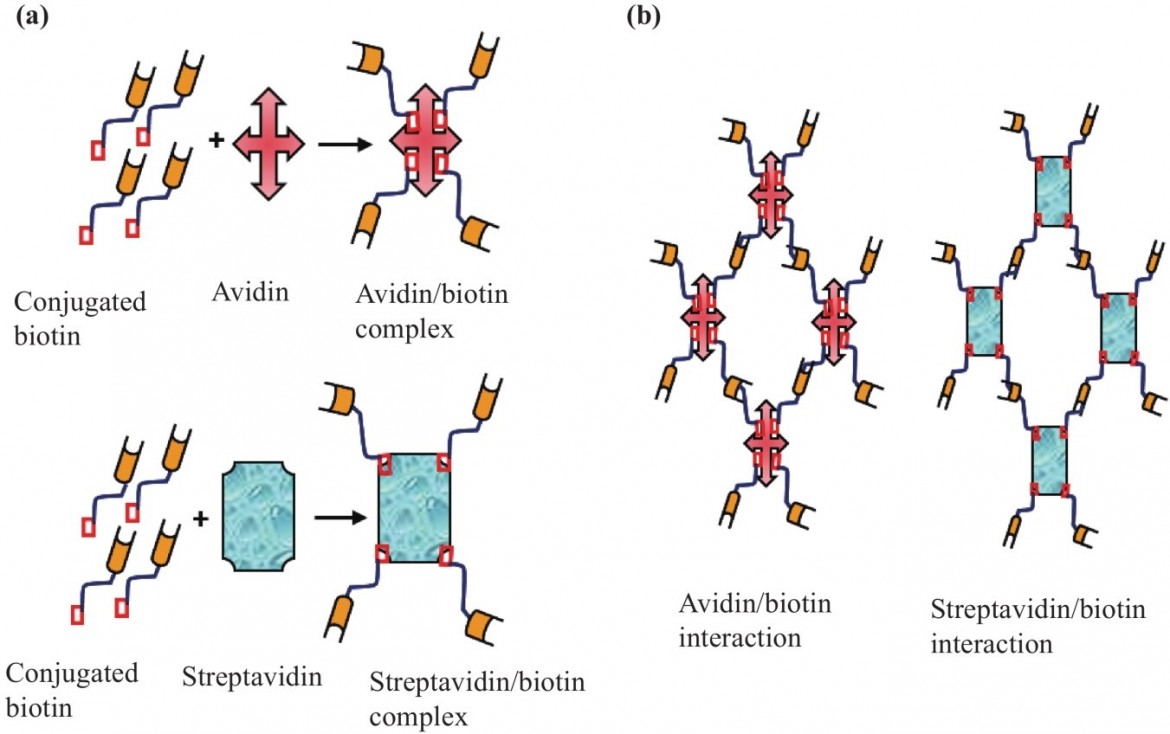
图4(a)亲和素/生物素和链霉抗生物素蛋白/生物素复合物的形成。
为了产生由此产生的抗生物素蛋白/生物素和链霉抗生物素素/生物素复合物,
抗生物素肽和链霉抗生素都可以与四种共轭生物素(通常与抗体、蛋白质、肽和酶共轭的生物素)分子结合。
(b)亲和素/生物素和链霉抗生物素蛋白/生物素之间的相互作用。
一种抗生物素蛋白/生物素复合物或链霉抗生物素肽/生物素复合体可以通过与共轭生物素结合
而与另一种抗免疫素蛋白/生物学复合物或链球菌抗生物素素蛋白/生化素复合物连接。
αvβ3-整合素在非活性内皮细胞或健康组织中缺失,是肿瘤新生血管内皮细胞的常见标志。在将抗αvβ3抗体直接注射到肿瘤中后,掺入了抗生物素蛋白和链霉抗生物素。这种磁性脂质体能够递送针对癌症细胞的特异性抗αvβ3抗体。在将磁性脂质体施用于靶向群体时,发现效应分子由于抗生物素蛋白和生物素之间的高亲和力而永久粘附在肿瘤预靶向抗体上。MRI显示肿瘤周围有很强的信号强度,占肿瘤面积的7%,而非靶向组仅改善了2%。根据组织学检查,磁性脂质体与新生血管系统共定位,导致磁共振信号减少。
4.2
配体锚定结合可以成功递送具有高特异性和靶位点区分的小分子药物。Avidin被证明是受体和配体之间的桥梁(配体可以进行功能修饰或未修饰)。因此,在一项研究中,发现通过用生物素紧紧封闭MSN的孔并用抗生物素蛋白覆盖MSN,可以防止顺铂从MSN的核心释放。将基质金属蛋白酶9(MMP9)特异性可切割接头添加到抗生物素蛋白封端的MSN中,该MSN在表达高水平MMP9的肿瘤中显示出位点选择性药物递送。可调的多孔结构和高容量使药物能够负载在这些NP中。使用这项技术,发现MMP9可以通过MSN刺激顺铂释放到人类肿瘤细胞中,以及在小鼠和人类肺癌的体外3D肺细胞培养中。此外,仅在Kras突变小鼠肺部释放顺铂的MMP9阳性肿瘤部位检测到凋亡细胞死亡。在小鼠的非肿瘤或未受影响的区域没有观察到毒性。
4.3 金纳米粒子
在另一项有趣的研究中,使用超分子方法通过链霉抗生物素蛋白(SA)-生物素结合与β-环糊精(β-CDs)和γ-环葡聚糖(γ-CDs)官能化来合成AuNP。值得注意的是,通过改变腔尺寸或官能化位点,可以合成许多不同的CD涂层NP。发现这些NP比天然NP更稳定。据观察NP@SA与γ-CD相比,γ-CD的DOX装载效率要高得多NP@SAβ-CD。相比之下,肾上腺髓质素(ADM)的负荷效率更高NP@SAβ-CD。因此,NP@SAβ-CD更适合装载较小的药物(ADM),而不是较大的药物(DOX)。
4.4 磷酸硅酸钙纳米粒子(CPNP)
CPNP通常通过增强渗透和保留(EPR)效应在实体瘤中积累。当这些NP与荧光染料结合时,其有效性可以显著增强。Barth等人将ICG引入掺杂硅酸盐的无定形磷酸钙基质中,以制备新型CPNP复合材料(直径约为20nm)。CPNP通过抗生物素蛋白-生物素偶联与生物素化二铁转铁蛋白(人全转铁蛋白)、生物素化抗CD71抗体(转铁蛋白受体特异性)和生物素化五肽胃泌素结合。生物素化人全转铁蛋白(二铁转铁蛋白)和生物素化抗-CD71抗体(抗转铁蛋白受体抗体)与含avidin-CPNP的相互作用允许更精确地靶向癌症细胞中丰富的转铁蛋白受体。因此,基于avidin-CPNP的治疗材料可用于治疗乳腺癌症和其他快速增殖和转移蛋白分泌的癌细胞。
4.5 碳酸盐磷灰石(CA)
最近,Mozar等人利用生物素-链亲和素相互作用制备了涂有亲水性和电中性PEG和纤连蛋白特异性配体的药物负载的CANP,以提高颗粒药物复合物通过受体介导的内吞作用(通过纤连蛋白和整联蛋白的相互作用)靶向和递送到乳腺癌症细胞中的能力。根据结果,CANP的平均粒径约为820nm,PEG-CA的平均粒直径减小到615nm,生物素化的PEG-纤维连接蛋白-CA的颗粒直径进一步减小到402nm。PEG-CA和PEG-纤维纤连蛋白CA的表面电荷比CANP更具正电性。在这项研究中,SA作为生物素化PEG和载药CA复合物之间的接头,因为CO32-和PO43-(带负电荷的结构域)与颗粒的富含Ca2+的结构域结合。因此,SA与生物素的相互作用促进了药物与NP的结合。使用高效液相色谱法(HPLC)评估癌症细胞对药物的摄取,结果表明表面修饰的NP的细胞摄取(疏水性药物)高于CA和游离药物。在细胞毒性研究中,表面修饰的NP比未修饰的CANP和游离药物显示出更大的实质毒性。此外,使用表面修饰的药物包封的NP进行的肿瘤回归研究显示,肿瘤显著缩小。因此,使用亲和相互作用进行表面修饰可以减小粒径,增加药物在肿瘤中的细胞内化和保留,并防止调理和脱靶分布。
4.6 碳纳米管
Bajaj等人使用拉曼成像免疫测定报告了一种生物素化的单壁碳纳米管(SWCNT)(通常直径为0.7-1.5nm,长度为10nm至几厘米),可以成功检测到共聚焦拉曼成像检测到的BT-474细胞中的Her2受体。发现单克隆Her-66抗体与Her2受体结合后,NeutrAvidin荧光素异硫氰酸酯(FITC)桥将生物素化的二抗和SWCNT连接起来。通过共聚焦拉曼显微镜和NeutrAvidin-FITC,通过BT-474细胞上产生的复合物通过免疫荧光观察Her2受体的表达。与非生物素化的SWCNT相比,生物素化SWCNT的拉曼信号降低了94%。这意味着生物素-亲和素相互作用可以减少生物素化SWCNT与第一抗体的非特异性结合。发现生物素化的纳米管通过Her2受体附着在细胞表面,而在细胞内的囊泡内观察到裸露的纳米管。正在研究使用带有SA功能化CNT/Ag-NP的一次性免疫传感器阵列进行超灵敏的多重肿瘤标志物检测。CNT/Ag-NP纳米杂化物是通过在羧化CNT上原位沉积Ag-NP而产生的。纳米杂化物通过蛋白质和银纳米粒子之间的内在亲和力用SA进行功能化,使信号抗体的生物素化能够获得标记抗体。通过官能化过程,纳米杂化物在水中的分散性得到了显著提高。捕获的抗体被共价固定在壳聚糖修饰的丝网印刷碳电极上,以创建免疫传感器阵列。
通过免疫传感器阵列上的三明治型免疫反应,将大量Ag-NP收集到每个免疫复合物上,在Ag-NP诱导Ag沉积后,Ag增强剂增强了免疫复合物溶液产生Ag-NP的电化学剥离信号。使用癌胚抗原(CEA)和甲胎蛋白(AFP)作为模型分析物,建议的多重免疫测定方法显示出令人满意的准确性和广泛的线性范围,检测限分别低至0.093和0.061pg·mL-1。采用所提出的方法,血清测定结果与参考值在可接受的范围内。新开发的方法和功能化标签消除了电化学免疫测定中的串扰和脱氧需求,表明它们可能在临床环境中有用。
5.通过共沉淀或阴离子交换反应(如LDH)插入NP
LDH是天然存在的合成层状材料,也称为阴离子粘土、类水滑石(HTI)或水滑石型(HTt)。大多数LDH材料的通式如下:x+(An−)·mHO(x=0.2-0.4;n=0.5-1),其中MII表示二价金属阳离子,MIII表示三价金属阳离子,An-表示阴离子,m表示溶剂的摩尔数(图5)。图5显示了用于治疗应用的LDH的结构。LDH是有效的药物载体,因为它们具有高度的生物相容性,具有双层结构,在热降解后可以恢复,易于制备,提供快速的药物递送,具有高载药量,并且对酶降解具有抗性。配体交换反应自由插层和表面修饰是将基因、核酸、药物、适配体、肽、抗体和蛋白质直接装载到LDH中的可行策略。肽、维生素、ATP和多糖等生物分子可以通过共沉淀或阴离子交换反应嵌入LDHs。与药物共沉淀提供了将LDH与药物结合的最精确和最直接的方法,因为它们不太倾向于引入CO32-和其他阴离子。所使用的药物必须能够在制备后程序(如水热处理)中存活,以提高所获得材料的均匀性和结晶度。然而,一些阴离子,如siRNA和反义寡核苷酸,无法抵抗这些条件。因此,他们使用阴离子交换法将其整合到LDH药物偶联物中。
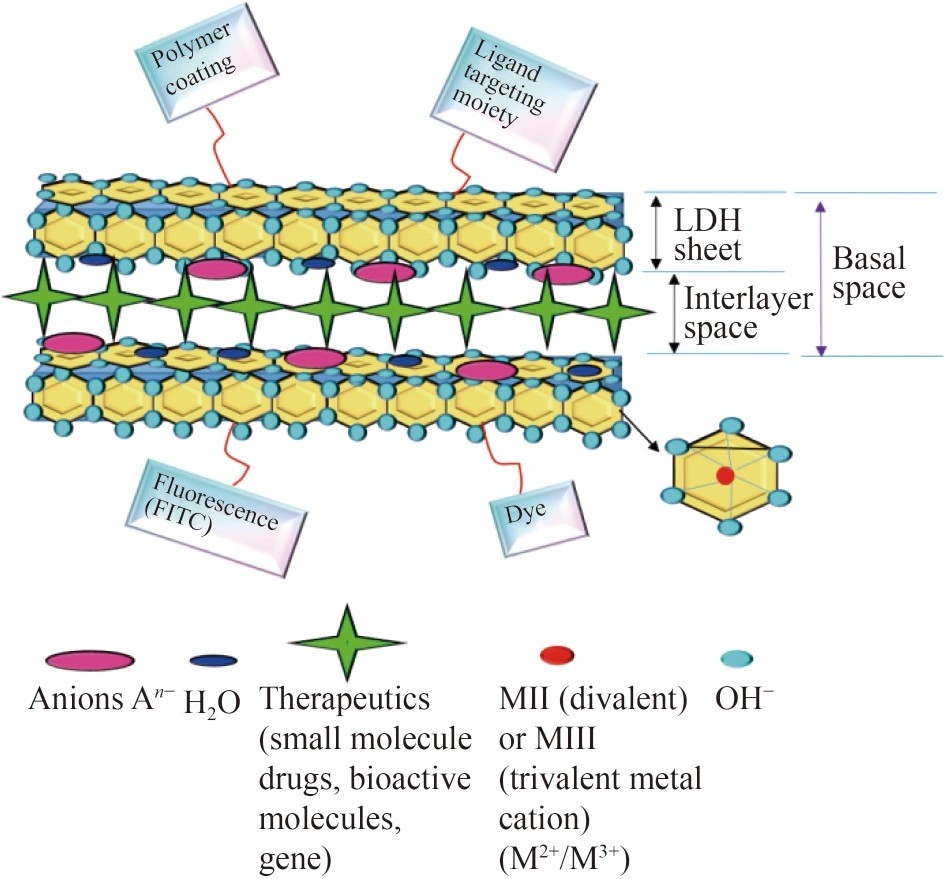
图5用于治疗目的的LDH结构示意图,其通式为x+(An-)x/n·mH2O(x=0.2-0.4;n=0.5-1)。
MII(M2+)表示二价金属阳离子,MIII(M3+)表示三价金属阳离子。
这里的“x”是M3+/(M2++M3+)的比率。M2+/M3+比率为2-4被认为相对稳定。
An−(在层间区域)是一种阴离子。An−代表任何电荷补偿的有机或无机阴离子,
例如含氧阴离子(碳酸盐、硝酸盐等)、卤化物、含氧阴离子和氧酸盐
(重铬酸盐,(Mo7O24)6−,(V10O28)6-等)m’表示不存在阴离子的层间区中所含溶剂的摩尔数。
LDH可以用无机离子、荧光(FITC)、染料、聚合物涂层和配体靶向部分进行修饰。
有证据表明,LDH可以有效地嵌入各种阴离子生物分子,如核苷酸、siRNA、DNA和抗癌药物,从而实现高生物利用度和有效性。为了最大限度地提高摄取效率,必须定义LDH的大小限制,以便用作细胞内载体。在一项研究中,使用流式细胞术评估了不同大小的LDH-FITCs的细胞摄取。研究发现,粒径对细胞吸收的LDH量有很大影响。最小的颗粒,50nm,很容易内化,其次是200≥100>350nm。只有极少量的350nmLDH可以渗透细胞,这一事实表明350nm太大,无法有效渗透细胞膜。所有这些发现都表明,尺寸在50至200nm之间的LDH通过网格蛋白介导的内吞作用选择性地进入细胞,而大于200nm的LDH则不能。因此,LDHs的大小依赖性摄取机制与摄取行为之间似乎存在很强的相关性。
NP容易产生尺寸依赖性毒性。一个研究小组调查了LDH对人类肺组织培养物的尺寸依赖性毒性作用。据透露,100-200nm尺寸范围内的LDH在细胞增殖、炎症反应和膜损伤方面表现出减轻的毒性。此外, 体内研究表明,不同大小的LDH不会影响体重或导致死亡率。因此,这些研究表明,LDH(在100-200nm范围内)可以促进生物相容性,增强药物的细胞内递送。大多数情况下,在LDH的内腔中,带负电荷的寡核苷酸通过离子交换反应嵌入。由于寡核苷酸嵌入LDH-NP中,负载的核苷酸受到保护,免受DNase攻击。
5.1 药物LDH偶联物
由于药物LDH偶联物的后续稳定性,药物的细胞内化可以得到改善,而不会产生任何明显的负面后果。其中一个例子是5-氟尿嘧啶-LDH(5-Fu-LDH),与游离5-Fu相比,5-Fu在肿瘤中表现出持续释放、延长半衰期和更高的积累。此外,5-Fu-LDH被证明是可生物降解的,给药后不一定在健康器官中积累。这表明它们已迅速从体内清除。Choy等人报告称,甲氨蝶呤(MTX)负载的LDH(MTX-LDH)可以通过网格蛋白介导的内吞作用促进细胞内化。然而,在共轭物的形态和功能性质方面没有观察到明显的变化。后来,Oh等人通过共沉淀法制备了类似的MTX-LDH。最终结果是,如3-(4,5-二甲基噻唑-2-基)-2,5-二苯基溴化四唑鎓(MTT)和5-溴-2-脱氧尿苷(BrdU)测定所证明的,在骨癌症细胞系(Sao-2和MG-63)中使用网格蛋白介导的内吞作用,增强了毒性和细胞内吞作用。
Choi及其同事使用带有HOS的异种移植物小鼠模型对完整MTX及其纳米杂交MTX-LDH的抗肿瘤作用进行了彻底的研究,其中48只小鼠分为四组:对照组(PBS缓冲液)、LDH(45mg·kg-1)、游离MTX(30mg·kg−1)和MTX-LDH(75mg·kg-1相当于30mg·kg-1MTX)。在第0、7和14天共静脉注射了三次。
MTX-LDH治疗的肿瘤比对照组小得多。需要注意的一点是,每个游离MTX治疗组和MTX-LDH组都接受了30mg·kg-1的MTX(~LD20值)。
Li等人调查的一项有趣的研究显示,芬布芬(FBF,一种用于缓解癌症疼痛的非甾体抗炎药)通过在氮介质中的共沉淀嵌入LDH-NP内部。X射线衍射(XRD)研究表明,与初始廊道高度(0.39nm)和水镁石状层(0.48nm厚)相比,在用FBF插层后,LDH层之间的距离增加到1.87nm。改变pH值对FBF-LDH插层有相当大的影响。当pH值从8增加到13时,FBF从单层变为双层,而距离的变化(1.87-3.00nm)指向嵌入结构。增加层间间距通过降低离子嵌入的扩散势垒和扩散能量势垒,增强了离子扩散动力学,特别是具有大尺寸和多价态的离子。基于Xue等人对紫杉醇LDH(PPT-LDH)修饰的研究,表明酪氨酸(Tyr)可以与LDH共沉淀以提高PPT的负载效率。因此,夹层空间被预先打开,为药物吸引创造了有利的环境。初步的体外抗癌研究证实了PPT-LDH杂化物对肿瘤细胞发育的抑制作用,载药效率为34%(药物/材料的w/w)。
5.2 配体LDH偶联物
在一项研究中,研究人员将靶向分子FA掺入LDHNP的表面,从而产生FA-LDH,以研究MTX-FA-LDH纳米杂化物在体外和体内模型中的靶向功能。除了EPR效应外,网格蛋白介导的内吞作用和叶酸受体介导的外吞作用被认为是提高摄取和治疗效果的原因。在一项siRNA递送研究中,KB细胞用siRNA转染或小鼠注射siRNA,发现siRNA(Survivin)-FA-LDH在体外显著降低了mRNA和蛋白质水平的基因表达,最终将肿瘤生长减轻了三倍,与体内没有任何靶向配体的SurvivinLDH相比。In从这样的结果来看,Survivin基因似乎比正常细胞和其他组织更有效地靶向肿瘤(1.2倍)。
癌症治疗通常需要两种或两种以上补充药物的化疗。然而,联合化疗尚未被证明是安全有效的。如Li及其同事所述,为结直肠癌癌症治疗设计了白蛋白稳定的LDH(BLDH)系统,以装载和递送两种常用的抗癌剂,5-Fu和白蛋白结合的紫杉醇(Abraxane,ABX)。168.3nm直径、0.181的聚合物分散指数(PDI)和14.0mV表面电位是所制备的BDLH/5-Fu-ABX系统的尺寸特性。BDLH/5-Fu-ABX的 体外药物释放模型表明,在酸性环境中缓慢的LDH中和可能导致5-Fu和ABX释放到癌症细胞的晚期内体/溶酶体中。在细胞摄取研究的基础上,BLDH/5-Fu-ABXNPs被结直肠癌癌症细胞(HCT-116)成功地内化,协同诱导癌症细胞凋亡。体内试验表明,BLDH/5-Fu-ABXNP在静脉注射三次后可以有效减少肿瘤的发展,且没有明显的副作用。治疗效果的显著提高归因于BLDH/5-Fu-ABX在肿瘤中的有效积累和酸敏感性共载药物的释放。因此,BLDH/5-Fu-ABXNP可能被用作治疗结直肠癌的新方法。
5.3 十二硼酸巯基十一氢酯(BSH)-LDH偶联物
Choy等人研究了使用硼中子捕获疗法(BNCT)输送硼的LDH纳米载体。需要大量的硼(B-10)才能成功地将BNCT递送到癌症细胞。阴离子硼分子簇,如美国食品和药物管理局(FDA)批准的B-10化合物BSH,与LDH插层以产生BSH-LDH,并最终被研究为硼递送系统。根据动物模型中涉及BSH-LDH纳米杂交药物(100nm)的生物分布研究,注射后2小时,BSH-LDH治疗组的BSH肿瘤与血液(T/B)比率比完整的BSH治疗组增加了4.4倍,从而支持了其通过EPR效应和网格蛋白介导的途径通过内吞作用靶向的机制。
5.4 无机离子LDH偶联物
对肿瘤酸性微环境有反应的纳米治疗平台对于准确的肿瘤识别和治疗至关重要。一个实验室报道了2D纳米治疗 平台的成功制造,其中功能性铁离子被掺杂到具有DOX的MgAl-LDH中以形成Fe-LDH/DOXNP,从而实现MRI引导的协同癌症化疗和光热治疗。将铁离子掺入Fe-LDH/DOX中会产生强烈的光热效应,转化效率为45.67%,与DOX结合使用可以协同杀死癌症细胞,同时进行光热治疗(PTT)和化疗。此外,T2加权MRI结果及其体外pH依赖性降解证明,Fe-LDH/DOX在肿瘤的酸性微环境中易感。重要的是,经PTT治疗和Fe-LDH/DOX化疗后,4T1携带小鼠的肿瘤生长得到了有效抑制。根据这些发现,将功能性金属离子掺杂到LDH-NP中可能会导致一种独特的方法,用于开发具有更高诊断和治疗能力的纳米治疗平台。
同样,Xu及其同事修饰了与pH敏感聚合物结合的带正电的含铜LDH-NP,以增强颗粒在血液循环过程中的胶体稳定性,防止健康细胞中的脱靶积聚,并促进肿瘤在肿瘤微环境中的分布和内化。基于体外实验,聚合物-LDH纳米载体降低了血液pH(7.4)中巨噬细胞的捕获,但由于聚合物涂层的脱离,在弱酸pH(6.8)中增强了癌症细胞的摄取。体内pH响应MRI验证了电荷可转换纳米杂化物在肿瘤中积累的能力,注射后24小时的积累率为4.8%(给药剂量),增强了它们作为抗癌纳米载体的多功能性。
有趣的是,LDH夹层能够替代任何有机或无机离子,无论是简单的还是复杂的。因此,LDH的稳定性取决于层间阴离子的性质。例如,Baek等人开发了Mg-al-LDH-Cl,在凋亡、膜损伤和氧化应激方面,其毒性低于Mg-al-LDH-CO3,因为它们在溶酶体的酸性环境中易于快速分解隔室(pH4.5)。
5.5 DNA-LDH偶联物
使用LDH进行快速基因表达/沉默涉及以下步骤:(1)负载DNA/siRNA的LDHs杂交体(50-250nm,总带正电荷)可以与带负电荷的细胞膜结合;(2)吸附后,LDH宿主可以通过网格蛋白介导的内吞途径内化到细胞质中,使其渗透到细胞膜中;(3)溶解LDH宿主后,DNA/siRNA在核处或核附近释放,在这种情况下,质粒将直接进入核,导致特定mRNA的表达或靶向,用于基因沉默。一个研究小组表明,FITC作为报告分子显著提高了用DNA/LDH杂化物处理的细胞的摄取,这种摄取随着细胞暴露于LDH的增加而成比例增加。结果表明,c-myc/LDH杂合物可被细胞快速吸收,反义寡核苷酸有助于细胞的代谢过程。将人白血病(HL-60)细胞暴露于AS-myc-LDH杂化物(20µmol·L−1浓度)4d后,癌症细胞增殖减少65%,表明LDH-NP能够将小核酸转移到靶细胞。此外,该小组报告称,随着时间的推移,细胞增殖受到抑制,并且呈剂量依赖性。
6.共价键合
共价附着具有通过附着化学调节药物释放的优势(例如通过GSH在细胞中释放基于巯基的有效载荷)。为了在药物和NP之间形成共价键,结合位点必须可用,并且没有强烈的静电排斥,并且具有微孔的极其致密和刚性的涂层。纳米载体上可用的各种官能团和待偶联的药物导致了共价偶联策略的发展(图6)。某些内部或外部刺激会触发共价结合的遗传物质的释放。siRNA可以共价连接到几种细胞穿透或靶向配体上,包括小分子受体、肽和脂质。然而,它们在血清中不稳定,循环时间短,阻碍了基因的长期沉默。碳纳米管、金NP和SPION通过共价功能化或巯基金共价化学结合小分子、siRNA和质粒。为了在肿瘤内诱导体液免疫反应,CNT可以作为抗原呈递载体,这可以改善基于肿瘤的肽和抗原的弱免疫原性。
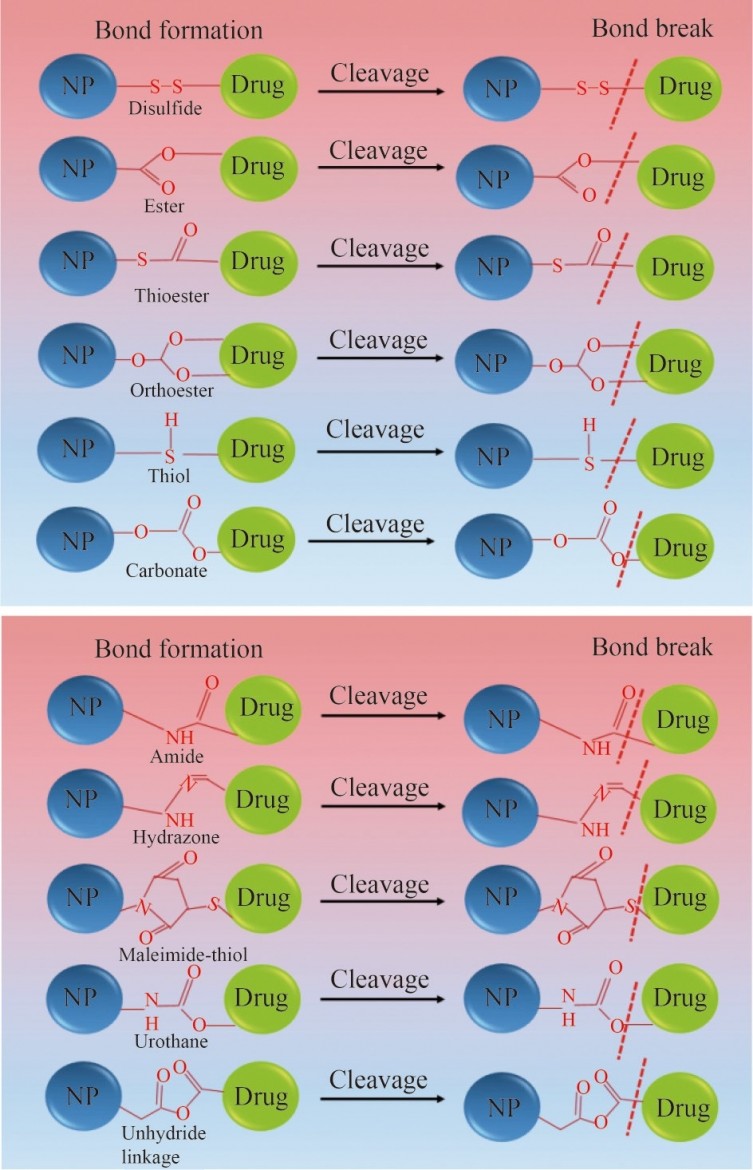
图6NP和药物之间共价键形成和断裂的示意图。所呈现的共价键主要基于纳米载体和共轭药物上可用的官能团
(例如,二硫化物、酯、硫酯、原酯、硫醇、碳酸盐、酰胺、腙、马来酰亚胺硫醇、尾烷和未氢化键)。
6.1 碳纳米管
化疗药物分子通常通过(1)与官能团的表面结合或(2)通过可切割的键产生CNT聚合物涂层与CNT结合。因此,生物分子、成像剂和药物可以通过用强酸(如HNO3和H2SO4)对其进行化学官能化而共价结合到CNT表面,这些强酸会引入碳纳米管尖端和缺陷位点的羧基和羟基等含氧基团,或通过1,3-偶极环加成反应,提供共价偶联的官能团。此外,使用CNT侧壁改性来引入不同的官能团允许纳米管的进一步衍生化。这些共价功能化技术主要用于修饰CNT以增强siRNA递送。
含有乙酰腙、酰胺或酯基团的共价键通常用于将药物结合到纳米载体或覆盖纳米载体的聚合物上。碳纳米管的聚乙二醇化最能说明它们在延长血液循环周期和减轻脱靶毒性方面的共价相互作用。例如,Liu等人证实,紫杉醇(PTX)可以通过可切割的酯键共价结合PEG包覆的SWCNT。SWCNT-PTX通过利用EPR效应,以比商业紫杉醇更大的效力减弱了4T1乳腺癌症细胞的增殖。因此,延长了血液循环的半衰期,并将PTX在肿瘤中的积聚增加了十倍。此外,SWCNT在RES中释放PTX,并通过胆汁排泄。Sobhani及其同事证明,与单独暴露于激光的组相比,在暴露于NIR激光10分钟后,瘤内注射PEG包覆的CNT可以减轻小鼠黑色素瘤肿瘤的生长。另一项类似的研究表明,与脾Treg或瘤内非Treg相比,在单次逆转录注射中与糖皮质激素诱导的肿瘤坏死因子受体(TNFR)相关蛋白(GITR)配体结合的聚乙二醇化SWCNT通过受体介导的内吞作用增强了B16黑色素瘤中调节性T细胞(Treg)的摄取,从而提高了对癌症的免疫反应。
6.2 金纳米粒子
巯基金共价化学是将siRNA偶联在金NP表面的整体方法之一。Nagasaki等人通过用巯基-PEG5000-PAMA7500聚合物修饰AuNP,然后自组装巯基化的siRNA,将siRNA偶联在AuNP表面。所得AuNP/siRNA偶联物对人肝癌HuH-7细胞显示出65%的基因沉默效率。为了提高DNA的细胞内递送,Lee等人在与聚β-氨基酯(PBAE)络合之前,通过二硫键用PEG修饰AuNP。TEM显微照片显示,PBAEsiRNAAuNP的核壳结构直径约为100nm。在与PBAE结合时,siRNAAuNP的表面电荷从-34mV变化到+13mV,这通过与带负电荷的细胞膜相互作用促进了细胞内递送。很明显,巯基金化学改善了siRNA向作用位点的递送,并促进了基因沉默的有效性。
Huh团队的研究人员开发了氧化铁/金纳米颗粒,并研究了它们的光活性和(体外和体内)生物活性。Au和S之间的共价连接使这些NP被巯基化的脱镁叶绿酸肝素包覆。在肿瘤细胞摄取后,谷胱甘肽通过消融Au-s键恢复了光敏剂的光活性。通过测量有或没有GSH的光敏剂的荧光来评估淬灭和去淬灭。孵育60分钟后,荧光强度(670nm)比没有谷胱甘肽的对照组增加了九倍。在9,10-二甲基蒽(1O2捕集器)存在的情况下,1O2的产生是明显的。共聚焦激光扫描显微镜用于检查A549细胞中的渗透,这揭示了内化NP的显著光敏剂信号,支持GSH在细胞质中的作用。无论NP是游离的还是释放了光敏剂,暴露在光下都会降低细胞存活率。在A459荷瘤裸鼠异种移植物模型中,游离光敏剂和NP之间的比较表明,NP比游离脱镁叶绿酸具有更高的肿瘤选择性和更长的肿瘤停留时间,而后者则迅速从体内清除。每2天给肿瘤诱导的小鼠注射生理盐水(对照组)、游离光敏剂或NP,并辅以光源。在检查肿瘤重量和体积时,很明显NP治疗组的表现优于游离光敏剂2.5倍。
另一种技术涉及使用触发链接器。Chen及其同事的研究应用了Au-S共价相互作用,将Au-NP偶联到Si-酞菁Pc4上。有趣的是,光敏性使这种配体与众不同,在近红外辐射(660nm)后,Si−C键发生溶血性光解,从而在配体与中间Si原子上的水交换后释放出Pc4。研究人员证明,PDT在HeLa中与非共价结合的Pc4一样有效但是共价相互作用促进了Pc4的更有针对性的释放,因为AuNP在运输过程中抑制了它。
寡核苷酸与Au核的共价连接导致寡核苷酸紧密地包裹在Au核周围,这在改变细胞对AuNP的反应方面具有明显的好处。这些发现也更普遍地应用于HeLa细胞,基于分析各种金NP的全基因组表达谱。使用寡核苷酸功能化的AuNP(ssDNA、dsNA和dsRNA),细胞在细胞周期调控、基因表达和凋亡方面的反应是适度的。相反,未经修饰的柠檬酸盐封端的金纳米颗粒被证明对细胞产生了明显的负面影响,包括凋亡的发生,这与柠檬酸盐封盖的弱附着有关,与牢固附着、密集寡核苷酸功能化的纳米颗粒相反。本研究强调了对表面功能进行稳健控制的重要性,这解释了共价相互作用的普遍性。
释放DNA的典型方法是使用光响应性NP,如纳米笼、纳米壳和纳米棒。可以调整这些NP的直径,以在NIR透明窗口中获得相当大的吸收。光热效应和适当频率的激光诱导电子产生可以诱导与这些NP共价连接的DNA释放。Chen等人使用这种在棒的纵向等离子体波长下照射飞秒激光的方法来诱导编码与Au纳米棒结合的增强型绿色荧光蛋白(EGFP)的质粒DNA的释放。这种方法使棒熔化,同时释放DNA,使共价附着在纳米棒表面的DNA分子释放高达80%。此外,细胞中EGFP荧光的存在表明质粒已成功转染。
对于铂(II)离子的细胞内释放,铂(IV)-NP复合物作为药物递送载体和前药引起了人们的关注。该方法具有优势,因为Pt(IV)与生物制剂的反应性低于Pt(II),从而降低了全身毒性并提高了生物利用度。Lippard等人采用传统的碳二亚胺偶联化学与1-乙基-3-(3-二甲氨基丙基)碳二亚胺和N-羟基琥珀酰亚胺,生成具有末端十二烷基胺部分的寡核苷酸官能化AuNP,与含羧基的Pt(IV)络合物连接。Pt(II)物种是通过含GSH的铂络合物中Pt(IV)的细胞内还原产生的。在失去轴向配体后,游离的顺铂将被释放。缀合物以与常规DNA官能化NP相同的方式被吸收到细胞中。研究表明,尽管大多数IC50值相似,但这些复合物在各种癌症细胞中具有与顺铂相似或等效的抗癌功效。Min及其同事采用类似的方法将Pt(IV)前药与Au纳米棒连接。在一端使用原位二硫代氨基甲酸盐生产,金纳米棒首先用二氨基(聚乙二醇)共价修饰,然后与含羧基的Pt(IV)分子(通过氨基)偶联。与游离顺铂相比,基于MTT测定的细胞毒性分析显示,三种独立的癌症细胞系的毒性明显更大,IC50值降低约9至65倍。这些结果与纳米棒处理细胞中Pt离子的较高细胞内浓度有关,这意味着NP递送系统可能通过不同的内吞途径在改善药物摄取方面发挥关键作用。
6.3 磁性NP
蛋白质和药物之间共价键的断裂是药物释放不可或缺的。这种结合可以在体循环中持续,也可以在肿瘤细胞内破裂并在靶点释放。例如,Ding等人证明,85%的DOX负载是通过(转铁蛋白)涂层的间接共轭实现的Fe3O4@SiO2NP通过多臂接头聚-L-谷氨酸(PLGA)。PLGA通过双功能磁性NP(DMPs)与转铁蛋白(Tf)和DOX化学偶联。经DLS和TEM证实,各NP的平均直径约为90.8nm,ζ电位为-44.1mV。这些数据表明它们的聚集倾向较小。值得注意的是,DOX通过酰胺键与NP的PLGA涂层连接。TfDMPNP证明了Tf的受体靶向功能,以及高DOX负载百分比、pH敏感药物释放和改善的Tf受体-抑制肿瘤细胞摄取,表明与未经Tf修饰的DOX偶联的DMP(DDMP)相比,其对癌症细胞具有更显著的细胞毒性作用。Kresse等人报道了体内Tf和SPION之间的共价相互作用在携带SMT/2A肿瘤的大鼠(大鼠乳腺癌)模型中成像。注射150分钟后,偶联物将MRI肿瘤信号降低了40%(范围为25%-55%),信号降低在正常大鼠中持续8小时,半衰期为17分钟。相反,使用相同的亲本SPION或用人血清白蛋白(HSA)标记的SPION,肿瘤信号仅降低了10%。
此外,研究人员使用药物和纳米载体之间的共价交联来靶向癌症细胞。例如,Yu等人通过共价交联设计了DOX负载的热交联(TCL)SPION,用于靶向递送和MRI。这个DOX@TCL-SPION其平均粒径为(21±6)nm(PDI=0.13),ζ电位为(-25±2)mV,比TCL-SPION(-37±2)mV更具电负性DOX@TCL-SPION验证了TCL-SPION上COO基团的一些负电荷被DOX(带正电)中和。体内研究表明,使用DOX@TCL-SPIONs与用5%葡萄糖、TCL-SPION、DOX(0.64mg·kg-1)和DOX(5mg·kg−1)治疗的小鼠相比。因此,DOX@TCL-SPION允许MRI被动靶向和检测肿瘤。此外,基于肿瘤内的药物积聚和随后从NP中释放,它们显示出增强的抗肿瘤疗效。
7.离子相互作用
为了构建能够将治疗剂和成像剂递送到肿瘤部位的纳米治疗平台,必须考虑药物和NP之间的离子相互作用。一般来说,用胺基、阳离子大分子(聚合物、脂质)或钙掺杂的CaP和CaCO3颗粒修饰NP表面以与小分子药物、siRNA和质粒络合的策略源于观察到许多药物递送平台表现出较差的治疗效果。这一概念证明了开发和优化纳米疗法以增强位点特异性药物输送的必要性。
钙掺杂、钙磷和碳酸钙颗粒与药物的相互作用CaP/CaCO3-NP的低毒性和增强的内体破坏能力使其成为DNA/miRNA/siRNA的有前景的载体。此外,钙离子上的正电荷为带负电荷的核酸提供了有用的结合位点,从而增强了它们对核糖核酸酶、RNA酶(siRNA和pDNA)核酸酶攻击的抵抗力或稳定性。大多数情况下,钙离子用于结合反义寡核苷酸中的磷酸骨架。
7.1.1 碳酸钙纳米颗粒
为了在肿瘤治疗中控制pH值,Som及其同事制备了一种单分散的非掺杂球霰石纳米CaCO3。研究人员观察到,在肿瘤上选择性沉积纳米碳酸钙会随着时间的推移提高肿瘤的酸度,从而阻止肿瘤生长。合成的NP的直径为20至300nm。在HT1080荷瘤小鼠中,静脉注射1mg推注剂量的纳米CaCO3使肿瘤pH值升高了3小时以上。100nm纳米CaCO3的pH值最高,影响持续时间最长。20nm颗粒比100nm颗粒更快地扩散进出肿瘤。此外,没有迹象表明300nm纳米CaCO3可以显著影响肿瘤的pH值。由于这些颗粒的扩散速率较低,它们只能穿透3D肿瘤组织的最小区域。因此,利用这项研究,有可能开发出一种pH敏感的纳米平台改变癌症的酸性环境。
同样,Kamba等人制备并测试了CaCO3纳米晶体,作为DOX的有效递送载体。这些纳米晶体以高水平的选择性和特异性诱导癌症细胞凋亡,而不会引起非特异性损伤。DOX插层到CaCO3纳米晶体中分别发生在高负载(4.8%)和包封水平(96%)下。CaCO3/DOX纳米晶体在生理pH值(7.4)下非常稳定,导致延迟释放。然而,在酸性pH值(4.8)下,纳米晶体解离更快,导致DOX释放更快。MDA-MB-231乳腺癌症细胞显示出对CaCO3/DOX纳米晶体的显著吸收,提供了潜在DOX递送的证据。使用MTT、改良的中性红/台盼蓝测定和LDH的体外化学敏感性试验证明,CaCO3/DOX纳米晶体比游离DOX更能抑制肿瘤细胞的生长。根据这些发现,CaCO3纳米晶体可能有助于控制输送药物和癌症的治疗。类似地,Hammadi等人创建了一种负载CaCO3的多烯紫杉醇(DTX)纳米载体,用于乳腺癌症筛查。为了评估产生的NP的药物递送和释放能力,在生理环境(pH7.4)和细胞内溶酶体环境(pH4.8)中对其进行了体外测试。物理化学研究成功制备了均匀的多形性纯文石型DTX-CaCO3-NP(平均粒径37nm),具有优异的结晶度和96%的包封率,在pH7.4下具有持续释放特性。制备的NP的平均直径在5至100nm的范围内,这使得它们不太可能被脾脏、肾脏和肝脏清除。在800–1000g·mL-1的浓度下,游离CaCO3NP的细胞存活率为90%,表明这些纳米晶体具有细胞相容性。24小时后,观察到合成的载药NP对MCF-7细胞的毒性作用比DTX低,而在48和72小时时获得了类似的作用,这与NP的缓释性能有关。因此,假设DTX的长效特性将提高生物利用度,减少剂量。
直径小于600nm的纳米载体可以通过利用肿瘤部位的EPR来改善药物在肿瘤中的内化。彭和研究小组使用多级自组装方法制备了pH敏感的介孔CaCO3。抗癌药物依托泊苷以稳定的形式包封,能够高效载药。如MTT法所示,与游离依托泊苷相比,产生的纳米结构更有效地抑制了SGC-7901细胞,并减轻了依托泊甙在HEK-293T细胞中的毒性作用。根据研究小组的说法,这些纳米载体在递送依托泊苷方面比游离依托泊甙更有效,对肿瘤细胞生长的抑制作用更强。为了响应外源性或细胞内刺激,通过EPR效应在肿瘤细胞中的NP分布,可以在比正常细胞低的pH值下诱导依托泊苷的快速稳定释放。因此,介孔CaCO3纳米球通过克服其不溶于水的性质,提供了一种增强依托泊苷等药物治疗效果的方法。
7.1.2 CaPNP
由CaP组成的载体结构的表面修饰可以促进治疗药物在肿瘤中的位点特异性递送和释放。一项研究旨在通过使用不对称的脂质双层稳定CaPNP来实现肿瘤靶向siRNA递送。此外,在人H460肺癌癌症异种移植物模型中,用PEG靶向茴香酰胺配体修饰的NP显示出显著改善体内基因沉默以及siRNA的靶向递送。与LPD(脂质/聚阳离子/DNA复合物)相比,LCP-II(脂质/钙/磷酸盐II型)表现出更大的曲率和更小的粒径,可以优先在肿瘤部位积聚治疗药物。此外,在H460异种移植物肿瘤模型中,通过单尾静脉注射含萤光素酶siRNA的NP后,检测到siRNA沉默活性(由LCP-II递送)。鉴于siRNA的成功递送,Wu等人开发了两种酶反应PEG/脂质/磷酸钙杂化物制剂(siRNA@NP1以及siRNA@NP2).据报道,几乎中性带电没有明显影响siRNA@NP2血清引起的分解。具体而言,siRNA@NP2交付效率高于siRNA@NP1,这不会引发酶反应。此外,体外和体内研究表明,NP相对安全。因此siRNA@NP2递送方法可能有助于基于siRNA的癌症基因治疗在体内的分布。
由于癌症(TNBC)的特点是DNA修复重和抗原表达阴性,化疗和内分泌治疗通常无效。快速解决这个问题对于改善治疗结果至关重要。Dong等人开发了一种基于CaP的pH敏感壳核平台,以协同方式向TNBC患者施用BRCA1siRNA和Pt前药(Pro-Pt)。在薄膜水合过程中,DSPE-PEG(1,2-二硬脂酰-sn-甘油-3-磷乙醇胺-聚乙二醇)自组装成含有亲脂性Pro-Pt的胶束,导致Pro-Pt在细胞内还原时转化为Pt,从而引发DNA损伤。胶束在其表面形成多孔壳,Ca2+和PO43-(CaP)通过电相互作用和物理吸附与带负电荷的siRNA结合。值得注意的是,发现CaP外壳在酸性溶酶体中分解,通过与溶酶体建立离子对,使溶酶体逃逸并连续释放膜。这确保了溶酶体的逃逸以及siRNA和Pro-Pt的顺序释放,阻断了BRCA1siRNA的DNA修复,并将Pro-Pt还原为Pt,导致不可逆的DNA损伤。此外,暴露的PEG亲水链产生了一种水合涂层,稳定了血液中的NP,防止它们被血清蛋白沉淀或被核酸酶灭活。此外,尿激酶纤溶酶原激活物类似物(uPA,对TNBC细胞和肿瘤相关成纤维细胞上高度表达的uPA受体具有高亲和力,但在正常组织中几乎找不到)是通过双重被动和主动肿瘤靶向能力将NP引入肿瘤细胞的极好介质。除了提供增强的包封效率和稳定性外,pH敏感的NP还抑制了siRNA和Pro-Pt在整个循环中的分解,确保了它们在肿瘤组织中的活性。
在邱及其同事的研究中,阿仑膦酸盐-透明质酸接枝聚合物(AHA)被涂覆在磷酸钙-siRNA共沉淀表面,以产生透明质酸(HA)官能化的磷酸钙NP(CaP-AHA/siRNANP),适用于将siRNA递送到靶组织。CaPAHA/siRNANP显示出均匀的球形几何形状,平均尺寸和表面电位分别约为170nm和-12mV。由于Ca2+和PO43-可以强烈相互作用,因此发现用亲水性HA涂覆NP可以在一个月内提高其物理稳定性。此外,体外研究表明,CaP-AHA/siRNANP能够通过CD44介导的内吞作用将靶向EGFR的siRNA递送到A549细胞中,并可显著降低EGFR表达。此外,siRNA从内部CaP-AHA/siRNANP中的pH依赖性释放表明,溶酶体酸化有助于NP的分解,导致内渗透压急剧升高,并显著加速siRNA释放到癌症细胞的胞质溶胶中。此外,体内肿瘤治疗表明,当将CaP-AHA/siEGFRNP静脉注射到移植了A549肿瘤的裸鼠体内时,肿瘤显著缩小,同时EGFR基因沉默,小鼠模型的体重减轻最小。这些发现表明,基于CaP-AHA/siRNANP的系统siRNA递送策略可能是靶向癌症细胞的有效和安全的选择。
7.1.3 CANP
最近,pH敏感的CANP被广泛探索用于有效递送药物、DNA、siRNA和蛋白质。我们的实验室证明,PEG对CA的表面修饰可以显著改善吉西他滨(一种亲水性核苷抑制剂)的递送和细胞毒性,提高乳腺癌症细胞的细胞内化,并延长药物-颗粒复合物的半衰期,这通过测量患有乳腺癌的小鼠血浆、癌症细胞和健康器官中吉西他宾的水平得到了验证。应该指出的是,改性的CANP是圆形的,看起来比未改性的CA-NP更坚固。据报道,在与吉西他滨结合后,CA的粒径急剧减小至685nm,表明吉西他滨具有减小粒径的潜力。当用没有或带有附加接头的生物素-PEG修饰时,负载吉西他滨的CA的粒径分别为537和554nm。蛋白质冠分析的数据表明,CA的聚乙二醇化会大大减少调理作用。一项针对乳腺肿瘤诱导小鼠的体内研究表明,与游离药物相比,吉西他滨在肿瘤中的积聚增加了近六倍,减少了健康组织中的脱靶分布。此外,有证据表明,基于高血浆药物浓度,聚乙二醇化颗粒的血液循环增加。因此,发现用PEG修饰CA可以成功提高其靶向药物递送的治疗效果。我们的研究小组还开发了负载AZ628的α-酮戊二酸修饰的CA,以减少NP的生长,并改善AZ628在4T1和MCF-7细胞系中的细胞内化和递送。在这两种细胞系中,AZ628负载的α-酮戊二酸修饰的CANP使细胞摄取增加了21%。因此,相应的NP可能有利于将AZ628递送至乳腺癌症细胞。此外,我们评估了柠檬酸盐修饰的CA和α-酮戊二酸修饰的CA在小鼠乳腺癌症模型中的抗肿瘤效果。与单独使用环磷酰胺(CYP)相比,用CYP负载的α-酮戊二酸修饰的CANP治疗的组显示肿瘤生长减少了五倍。此外,负载CYP的NP在肿瘤中的积累比游离CYP更多。生物分布研究表明,与游离DOX相比,DOX负载的NP在心脏中的积聚较少,表明它们在减轻小鼠心脏毒性方面的有效性。这些发现表明柠檬酸修饰的CA和α-酮戊二酸修饰的CA载体都可以延长循环时间,增强抗肿瘤作用,减轻化疗药物在健康组织中的毒性。在我们小组之前的一项研究中,Hossain等人证明,柠檬酸盐和琥珀酸盐修饰的CANP可以在MCF-7细胞中诱导更有效的DOX递送。与琥珀酸修饰的CA相比,柠檬酸修饰的CA对DOX结合的亲和力最强。此外,柠檬酸修饰的CA改善了MCF-7细胞的细胞内化,并具有半最大抑制浓度,比游离DOX低1000倍。因此,所有这些结果表明,pH敏感CA的药代动力学特征的改善(通过表面修饰)可能为NP药物递送的增强提供了一个连贯的解释。
7.1.4 羟基磷灰石纳米颗粒
一项关于柠檬酸盐改性羟基磷灰石(Cit-HA)NP表面功能化的研究强调了DOX在癌症细胞中的转运。由于DOX的正电荷和Cit-HA的负电荷之间的静电相互作用,DOX的封装效率提高了约85%(使用DOX:Cit-HA比=1:10)。在溶血和细胞毒性研究中,Cit-HA-NP显示出显著的低毒性和改善的细胞摄取(由荧光显微照片证实)。同样,Rodríguez-Ruiz等人在存在(cAp)或不存在(Ap)碳酸根离子的情况下开发了柠檬酸盐官能化的纳米晶磷灰石(通过亚稳态钙/柠檬酸盐/磷酸盐溶液的热分解制备),以装载和输送DOX。在7天后的生理pH值下,cAp-DOX释放的DOX比Ap-DOX多约42%。有趣的是,在酸性pH条件下,两种方法都产生了相似的DOX释放曲线。体外研究显示,cAp-DOX具有选择性内化、积累GTL-16人癌细胞并产生显著毒性的潜力。此外,发现静脉注射cAp-DOX在靶向肿瘤和延长血液循环时间方面更有效。
7.2 药物、siRNA和质粒与阳离子聚合物修饰的无机NP的相互作用
在癌症治疗中,用带正电荷的聚合物包裹NP可以改善NP的内在化。用阳离子聚合物包覆亲水性分子,如药物、DNA和肽,有望提高封装效率并抑制多核苷酸的酶降解。
7.2.1 与阳离子PEI改性NP的相互作用
一种特殊的阳离子聚合物是PEI,由于其内体活性和DNA缩合能力,主要用于修饰无机NP。此外,PEI在传递基因方面发挥了至关重要的作用,因为它们具有独特的缓冲能力(质子海绵效应),可以离开内体隔室。此外,PEI的高正电荷密度使细胞膜的负电荷成分能够 与PEI静电相互作用,从而通过内吞作用实现有效的基因递送。然而,研究表明,阳离子PEI的不同传输机制与质子海绵效应的假设相矛盾。已经开发了各种方法来将PEI分子接枝或物理吸收到无机NP的表面或内部。用PEI修饰的NP具有许多潜在功能,能够同时递送小分子药物、核酸(DNA、RNA和寡核苷酸)和成像剂,从而实现有效的成像引导协同药物和基因治疗。有趣的是,通过这种方法,基因治疗可以变得更安全、更有效。
7.2.1.1
与PEIhase结合的无机NP在改善癌症治疗方面引发了许多研究兴趣。由于PEI具有分支结构,因此可以进行简单的表面修饰,从而使其成为siRNA的合适载体(体外和体内)。据报道,在一项研究中,使用静电吸附过程将siRNA负载并保护在用超支化聚合PEI(hbPEI)修饰的MSN孔内。与未修饰的MSNs相比,PEI包覆的siRNA/MSN更有效地递送siRNA,并增强了细胞中的siRNA摄取。
Li等人建立了具有外部PEI表面涂层的负载siRNA的磁性MSNs(M-MSN),以及化学偶联的融合原性KALA肽(扭曲为M-MSN_siRNA@PEI-KALA).观察到NP的细胞毒性较低。因此,NP很容易渗透细胞,从内溶酶体中逃逸,并在细胞质中释放siRNA。瘤内注射M-MSN_VEGFsiRNA@PEI-KALA在体内阻止癌症细胞生长,可能通过抑制肿瘤新生血管。为了增加分布、保护siRNA并将siRNA递送到特定位点,构建了一种新一代siRNA递送系统,该系统具有PEI包覆的MSN核心,然后用PEG和抗体包覆。该构建体包括一个47nm的MSN核心,包裹在交联的聚乙烯亚胺-聚乙二醇共聚物中,该共聚物含有靶向HER2癌基因的siRNA,与曲妥珠单抗(抗HER2单克隆抗体)结合使用。该构建体旨在延长siRNA在循环中的半衰期,改善肿瘤细胞的摄取,并增强siRNA的敲除。然而,在HER2+乳腺癌症细胞中,靶向抗HER2NP触发细胞死亡,而在HER2-乳腺癌症细胞中,它们不触发细胞死亡。在曲妥珠单抗耐药的HCC1954异种移植物中,单剂量siHER2NP使HER2水平降低了60%。在三周的时间里,发现多次静脉注射可以显著减少肿瘤增殖。当暴露于人类外周血单核细胞时,siHER2NP在血液相容性和最小细胞因子产生方面表现出出色的安全性。该结构在批量生产中高度一致,制造程序有利于大规模生产。显然,这些siHER2NP可以进行临床评估。
Shen及其同事提出了一种可行的策略,可以最大限度地减少与未改性PEI相关的电荷诱导毒性,其中用PEI官能化的MSN孔接枝在环糊精(CD)上。使用荧光标记的siRNA和体内成像证明了CD-PEI-MSN在MDA-MB-231乳腺癌移植小鼠模型中释放siRNA的功效。在靶细胞中处理6小时后观察到弥漫荧光,可能是由于“质子海绵效应”导致siRNA从内体释放。“质子海绵效应”是由多价阳离子表面的缓冲能力引起的,导致盐流入内体以调节正确的pH值,最终破坏内体膜并将siRNA释放到细胞质中。
7.2.1.2 氧化铁NP
Tutuianu及其同事合成了PEI包覆的氧化铁NP(Fe-PEI),以研究其与顺铂的抗肿瘤作用。结果表明,与游离顺铂相比,负载顺铂的Fe-PEINP可以更有效地抑制癌症细胞生长(体外和体内)。一项类似的研究报告称,用阳离子烷基化PEI(Alkyl-PEI2k)涂覆磁性氧化铁(MIO)纳米簇会产生一种生物相容性复合物,该复合物能有效递送siRNA,并抑制4T1细胞和fLuc-4T1异种移植物模型中萤光素酶基因的表达。
Li等人制造了PEI功能化的氧化铁介孔二氧化硅卵黄壳纳米胶囊(NC),该胶囊通过与PEI的相互作用将siRNA静电附着在表面。该车的“蛋黄壳”功能集成了多种材料,并利用了它们的独特品质。介孔二氧化硅壳的荧光和氧化铁蛋黄的顺磁性将允许荧光成像和磁导航。这个PEI-Fe3O4@fmSiO2在外部磁场的引导下,卵黄壳NCs被证明可以有效降低HeLa细胞中的β-肌动蛋白表达,而不会表现出PEI的典型毒性。
7.2.1.3 碳纳米管
为了降低毒性并延长血液循环时间,CNT也用PEI进行了功能化。一个例子是用琥珀酰化聚乙烯亚胺(PEI-SA)对SWCNT进行非共价功能化,该物质在携带黑色素瘤的C57BL/6小鼠中递送siRNA。发现荧光Cy3标记的Braf特异性siRNA(siBraf)的基因沉默和显著的细胞内化。此外,IS/C/siBraf的25天应用导致肿瘤体积减小。Wu等人将多壁碳纳米管(MWCNT)与PEI接枝,然后与异硫氰酸荧光素(FITC)和前列腺干细胞抗原(PSCA)单克隆抗体(mAb)结合。CNT-PEI(FITC)-mAb复合物在体外和体内测试时表现出改善的生物相容性。此外,超声(US)成像验证了它们作为造影剂的能力。在PC-3荷瘤小鼠中,发现CNT-PEI(FITC)-mAb显著抑制肿瘤生长,表明其作为造影剂和药物载体的巨大潜力。
7.2.1.4 金纳米粒子
特别是在癌症治疗中,迫切需要纳米级基因递送载体,用于分发siRNA。Lee等人领导的研究小组证明了利用邻苯二酚偶联的PEI(PEI-C)进行siRNA递送来控制PEI包覆的AuNP的生产。在水性条件下,PEI-C产生了球形多核胶束,由于共轭邻苯二酚基团的还原性和疏水性,其作为还原模板用于形成和制备具有可定制粒径和表面电位的球形金纳米颗粒。通过交联,PEI-C牢固地附着在结晶金晶种的表面,形成阳离子金NP。由于PEI涂覆在AuNP上,形成了稳定的siRNA-AuNP复合物,这对抑制肿瘤细胞上的基因表达具有深远的影响。观察到内化和siRNA解包以及基因沉默的有效性在很大程度上取决于金NP的粒径和表面电位。由于伯胺基团密度低,水中缺乏未复合的PEI部分,PEI包覆的AuNPs的细胞毒性极低。为了更好地了解粒径对细胞转染效率的影响,将AuNP与PEI偶联,得到两组PEI包覆的AuNP,其粒径分别集中在约6nm(<10nmAuPEINP)和70nm(<100nmAuPEINPs)。在AuPEINPs/DNA复合物制备过程中,编码报告基因或自杀基因的质粒附着在AuPEINP上。使用人骨肉瘤Saos-2细胞来评估Au-PEI-NPs在富含血清的培养基中作为转染载体的能力。发现Au-PEI-NP两种类型在DNA偶联物中都带负电荷。尽管质粒结合的NP和细胞表面之间存在静电排斥,但两种类型的Au-PEI-NPs都出现了细胞摄取。由<10nm的Au-PEI-NPs形成的复合物很好地转染了细胞;然而,来源于<100nmAu-PEI-NPs的复合物则没有。较小的Au-PEI-NPs作为转染载体的功效与它们在细胞中的聚集减少和DNA从内体逃逸有关。同时,在与<100nmAuPEINPs孵育的内吞囊泡中鉴定出与DNA相关的大团NPs。
7.2.1.5 氧化石墨烯(GO)NP
siRNA沉默特定蛋白的事实表明,它可以显著降低癌症细胞的MDR。在最显著的抗凋亡防御蛋白中,Bcl-2与癌症细胞的MDR密切相关。使用Bcl-2靶向siRNA抑制癌症细胞中Bcl-2蛋白的表达,将成功对抗癌症细胞的MDR,并使其得到更有效的治疗。Zhang等人通过用氧化石墨烯(PEI-GO)官能化PEI,以顺序方式递送Bcl-2靶向siRNA和抗癌药物DOX。据报道,PEI-GO是递送siRNA和小分子药物的理想纳米载体。此外,PEI-GO将siRNA和DOX顺序输送到癌症细胞中产生了协同效应,从而显著提高了化疗疗效。
7.2.2 阳离子壳聚糖改性纳米颗粒的相互作用
在许多治疗用途中,壳聚糖(一种具有来源于壳聚糖的天然胺基的聚合物)是癌症治疗的有吸引力的候选物。鉴于其内在特性(如生物相容性、结构变异性、粘膜粘附性、生物降解性、无毒性和静电效应,使其易于与DNA等阴离子生物大分子结合),许多研究都集中在将其与无机载体一起使用上。
7.2.2.1
Gurka等人关于壳聚糖对MSNs和尿激酶纤溶酶原激活物配体(UPA)功能化的研究强调了它们作为pH敏感纳米载体的有效性,将药物输送到肿瘤的酸性微环境。在本研究中,壳聚糖用于提高药物负载效率,UPA作为纤溶酶原激活物受体在胰腺癌症细胞中过表达。
Murugan等人通过在MSN表面负载与槲皮素(QT)偶联的拓扑替康(TPT)药物,开发了一种纳米载体。用聚(丙烯酸)(PAA)和壳聚糖包被药物负载的MSN,通过整合素受体介导的内吞作用,将MSN与精氨酸-甘氨酸-天冬氨酸(cRGD)连接以靶向癌症细胞,尤其是癌症细胞。在这项研究中,由于组织间/细胞内pH值的变化,壳聚糖的分解引发了药物释放。
“肿瘤触发靶向”的方法允许开发基于壳聚糖的双pH敏感抗癌纳米载体(CHI/MSN),以提高药物疗效并最大限度地减少不良反应。MSN负载DOX并用苯并咪唑(Bz)修饰。接下来,作为“看门人”,壳聚糖接枝-β-环糊精(CHI-g-CD)通过β-CD和Bz之间的主客体相互作用包覆MSN。甲氧基聚乙二醇苯甲醛(mPEG-CHO)通过pH敏感的苯甲酰亚胺键接枝到CHI上,随后用靶向金刚烷-甘氨酸-精氨酸-甘氨酸-天冬氨酸丝氨酸(Ad-GRGDS)肽包覆。由于PEG在pH7.4时提供动态保护,因此所得载体被认为是“隐形的”,能够在弱酸性条件下暴露屏蔽的靶向肽和CHI的正电荷,从而实现“肿瘤触发靶向”。肿瘤细胞内β-CD和Bz组之间的关联可能会因pH值降低而中断,导致DOX释放。将携带肿瘤的BALB/c小鼠随机分为五组,DOX@MSN-CHI-RGD,DOX@MSN-CHI-PEG,以及DOX@MSN-CHI-RGD-PEG分别在第0、2和4天三次。这个DOX@MSN-CHI-RGD-PEG含有靶向肽比含有靶向多肽更有效地抑制肿瘤生长DOX@MSN-CHI-PEG独自一人。老鼠在DOX@MSN-CHI-RGD-PEG与PBS组相比,各组显示出最佳的肿瘤缩小效果,平均肿瘤重量约为18.0%。这个DOX@MSN-CHI-RGD-PEG在体外和体内研究中均显示可促进癌症细胞凋亡,减少肿瘤发展,并降低DOX对正常细胞的细胞毒性。因此DOX@MSN-CHI-RGD-PEG该系统被认为是一种可行的癌症治疗方案。
7.2.2.2 硒NP
由于TNF-α的半衰期短和致命的副作用,其治疗用途被显著减少。Yan及其同事构建了一种名为TNF-α衍生多肽(P16)偶联、壳聚糖(CTS)修饰的硒NP(SC)的稳定纳米药物,记为SCP,其中SC作为缓释载体附着在P16上。SCP减少了几种类型肿瘤细胞的增殖,包括DU145前列腺癌症细胞。然而,RWPE-1人前列腺上皮细胞不受SCP的抑制。SCP在诱导DU145细胞G0/G1期细胞周期阻滞和凋亡方面比P16和TNF-α更有效。在携带DU145肿瘤的异种移植物模型中,SCP表现出比P16或Estmustine(一种癌症治疗药物)显著更大的抗癌作用,不良副作用更少。此外,在DU145异种移植物肿瘤中,SCP抑制细胞生长并增加凋亡。此外,正如机制研究所证明的那样,SCP的抗癌作用是通过p38MAPK/JNK通路激活介导的,导致G0/G1细胞周期停滞,以及胱天蛋白酶反应性细胞死亡。根据这些发现,SCP可能是治疗癌症的可行方法。
7.2.2.3 硫化锌(ZnS)NP
对于口腔上皮癌的体内成像,Jayasree等人的研究采用了甘露糖官能化的壳聚糖硫化锌:锰(CS-M-ZnS)。物理化学稳定的掺杂ZnS纳米晶体因其物理化学稳定性、高度可调的光谱和极其稳定的荧光而被开发为有利于细胞的纳米复合物,在癌症区域具有优异的吸收效率,这优于其他重金属。发现生物偶联颗粒足够稳定,平均粒径约为150nm。壳聚糖改善了纳米晶体的生物相容性,并提供了足够的甘露糖基化功能。对小鼠成纤维细胞(L929)和口腔上皮癌(KB)细胞的体外细胞毒性测试验证了它们的细胞相容性。正如KB细胞过表达甘露糖受体所揭示的那样,甘露糖生物偶联赋予了特异性和选择性细胞标记特性。这些纳米晶体与CS-M的结合带来了许多好处,包括钝化荧光纳米晶体的表面缺陷,增加荧光发射,增强癌症细胞的内在化,改善了癌症成像和人类血清中的生物相容性,有可能改善癌症的诊断和预后。
7.2.2.4 金纳米粒子
如Manivasagan等人的一项研究所示,紫杉醇负载的巯基壳聚糖层状二氧化硅/金纳米壳与抗表皮生长因子受体抗体(抗EGFR-PTX-TCS-GNS)结合,被确立为荧光/光声双模的治疗指标成像引导的化光热协同治疗。合成的纳米复合物为152nm,表面电位为49mV。这种纳米复合物具有生物降解性、血清的高稳定性、生物安全性、延长循环、高近红外吸收、优异的光热稳定性、增强的药物包埋性、有效的靶向性以及暴露于激光时癌症部位药物的可控释放等特性,为化学光热治疗与光声成像(PAI)联合过程中近红外激光触发药物释放提供了理想的平台。在酸性环境中,纳米复合物在暴露于808nm的近红外激光48小时后释放药物的速度是正常条件下的三倍。抗EGFRPTXTCSGNSs+激光照射组导致81.9%的细胞凋亡。相比之下,游离PTX、PTX-TCSGNSs和抗EGFRPTX-TCS-GNS的光热治疗效能分别为25.45%、17.15%和13.27%。与所有其他组相比,抗EGFRPTXTCSGNS+激光照射组在34天内的存活率为100%。此外,用纳米复合物治疗5小时后,抗体修饰的缀合物内化约为非靶向样品的四倍,表明抗EGFRPTX-TCSGNS通过受体高度内化。在携带乳腺肿瘤的裸鼠中静脉注射纳米复合物显示,6小时后,由于强烈的近红外吸收和密集的血管,肿瘤处的血红蛋白水平PA信号(532nm)升高,显示了纳米复合物帮助肿瘤检测的令人印象深刻的能力。
7.2.2.5 钙磷NP
在最近的一项研究中,设计了一种siRNA-负载壳聚糖-磷酸钙NP(CS/CaP/siRNA-NP),作为治疗宫颈癌症的一部分。值得注意的是,纳米沉淀方法用于产生CS/CaP/siRNANP。所得NP为均匀球形,直径约为194nm,ζ电位约为+27mV。已经证明,使用这些NP可以将siEGFR连续递送到Hela细胞中,导致EGFR表达急剧减少,这可能是由于壳聚糖的细胞粘附性得到改善,从而为细胞摄取提供了更多时间。内化的CS/CaP/siRNANP随后显示出pH响应性NP分解,导致siRNA释放增加,溶酶体快速逃逸到细胞质中。此外,体内抗癌研究表明,在Hela肿瘤诱导的裸鼠模型中,CS/CaP/siRNANP在瘤内注射后可以有效抑制肿瘤生长,导致整个实验过程中体重没有显著差异。由于这些发现,CS/CaP/siRNANP为宫颈癌症治疗中siRNA的粘膜递送提供了巨大的潜力。
在另一项研究中,Roy等人使用藻酸盐包裹的壳聚糖包覆的磷酸钙(AEC-CP) 纳米载体来包裹Fe3O4bLf(Fe3O4饱和乳铁蛋白)(NC)。使用结肠癌癌症干细胞的异种移植物,将纳米制剂口服给注射三阳性(EpCAM、CD133、CD44)分选的结肠癌癌症干细胞的小鼠。在喂食非靶向(NT)NCs的小鼠中发现70%的肿瘤消退,30%的小鼠在治疗30天后出现癌症复发。然而,仅在10%的给予Tar(AEC-CP-Fe3O4-bLf)NC的小鼠中观察到肿瘤的复发,导致生存率显著提高。
7.3 与阳离子脂质修饰的纳米颗粒的相互作用
阳离子脂质是带有正电荷的两亲性化合物,在水溶液中自发形成带正电荷的脂质体,偶尔与中性辅助脂质结合。这些脂质体主要用于基因治疗或免疫试验,作为DNA、RNA或蛋白质的有效载体。与中性或带负电荷的脂质体相反,阳离子脂质体可以主要通过静电吸引与带负电荷分子(例如质粒、mRNA、核酸、蛋白质、寡核苷酸和肽)有效相互作用,以促进细胞摄取,促进核酸递送和治疗药物在所需部位的释放。此外,这些治疗性脂质体可以包埋无机NP,并可以将核酸治疗剂或药物掺入亲水性核心内,或将其嵌入外部的亲脂性双层中。图7显示了药物或核酸偶联的NP在脂质体中的掺入。
到目前为止,递送siRNA的最突出的基于脂质的纳米载体是阳离子脂质体。带负电荷的siRNA与阳离子脂质主要负责将siRNA装载到阳离子脂质体中。阳离子脂质有助于细胞内的渗透,并促进颗粒从内体中逃逸。一些阳离子脂质体,如1,2-二油酰基-3-三甲基丙烷铵(DOTAP)siRNA、N-(1--丙基)-N,N,N-三甲基氯化铵-siRNA和Lipofectamine®2000(美国马里兰州生命技术公司)-siRNA复合物,经常被用作siRNA的递送载体。然而,这些复合物通常在其表面具有显著的电荷密度,使其易于与血清蛋白相互作用并引发免疫反应,导致其迅速从血液中去除。
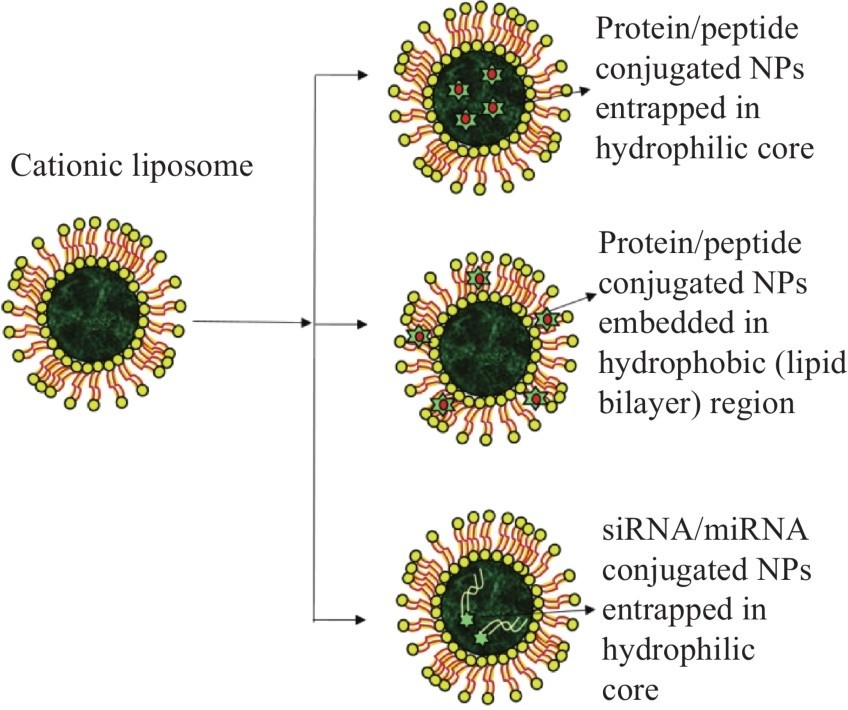
图7脂质体中药物或核酸偶联NP的掺入。
阳离子脂质体可以将负载无机NP的治疗剂包埋在亲水性核心内,或将其嵌入外部的亲脂性双层中。
7.3.1 CaPNP
许多研究表明,阳离子脂质结合无机药物递送平台可以有效治疗各种癌症。例如,涂有一层阳离子脂质(DOTAP(±)-N,N,N-三甲基-2,3-双(z-十八烷-9-烯酰氧基)-1-丙胺氯化物)的纳米级CaP核,然后用PEG和茴香酰胺(AA)配体接枝表面,在粒径和团聚控制方面发挥了重要作用。结果,实现了对表达σ受体的B16F10黑色素瘤细胞的高靶向性。各NP的平均粒径为40nm,表面电位为25mV。发现CaP核心在较低pH值的内体中分解并释放包埋的siRNA,而PEG涂层通过保护遗传物质免受核酸酶攻击,提高了体外和体内的基因沉默活性。在携带转移性肺肿瘤的C57BL/6小鼠中,单次静脉注射负载抗萤光素酶siRNA(0.12mgsiRNA/kg)的脂质磷酸钙(LCP)NP可将萤光素酶基因的转录降低78%。有趣的是,当针对靶向LCPNP中的MDM2(负调控p53肿瘤抑制蛋白的小鼠双分钟2-癌基因)、c-myc和血管内皮生长因子(VEGF)配制siRNA时,转移结节中的癌基因被沉默。在治疗过程中,在相对较低的剂量(0.36mg·kg-1)下,各自负载siRNA的NP显著减少了70%至80%的肺转移。相比之下,对照组没有明显效果。此外,这些靶向LCPNP是无毒的,与对照组相比,动物的平均存活时间提高了27.8%。Reinhardt等人开发了通过化学表面官能化暴露季铵基团(NPQ+)的二氧化硅NP,并根据漫反射红外傅里叶变换(DRIFT)、1HNMR光谱、ζ电位和光散射测量(胶体稳定性分析)对其进行了评估。共沉淀法用于证明在碱性pH下结合NPQ+的DNA递送的增强功效。冷冻电镜(Cryo-EM)图像表明,在NPQ+/DNA/阳离子脂质三元复合物中,DNA链夹在NPQ+表面和阳离子脂质双层之间。NPQ+不同寻常的静电胶体稳定性和在高盐浓度下增强的DNA结合亲和力导致了三元组装的发展,该组装可以调节这些复合物在生理环境中的稳定性和递送特性。
7.3.2 量子点(QD)
Al-Jamal及其同事研究了新设计的含有脂质体和量子点(L-QD)的囊泡在裸鼠全身治疗后的药代动力学。通过在各种双层组合物中添加疏水性量子点后测量浊度和羧基荧光素释放来评估杂交囊泡的血清稳定性。此外,还分析了元素(镉),以确定血液特征和LQD杂交种的组织积累。基于L-QD脂质双层特性,静脉注射后检测到不同的组织分布模式和亲和力。研究发现,阳离子(DOTAP/DOPE/Chol)杂化物在肺组织中容易积聚的同时,会迅速从血液中清除,但在两性离子囊泡的表面添加PEG会大大延长其在全身分布后的血液循环半衰期。总体而言,L-QD杂化囊泡系统被认为是一个有前景的平台,用于将QD分布到各种组织中,因为杂化囊泡的特性可以很容易地调节。此外,通过在单个囊泡的不同隔室中结合药物和QD,L-QD为开发双重治疗和成像(治疗-诊断)方法开辟了多种可能性。同一研究小组还通过将最小尺寸(2nm核心直径)的CdSe/ZnS量子点掺入阳离子1,2-二油酰基-3-三甲基铵-丙烷脂质双层中,自组装成小单层囊泡,证明了L-QD双层囊泡的制备。根据低温电子显微镜对含有L-QD的杂化双层的结构表征,QD的包含是由于疏水分子在生物膜内的自结合。在肺上皮细胞(A549)中,发现L-QD囊泡附着并内化,共聚焦激光扫描显微镜检测到细胞内运输的证据。此外,在人类癌症(C33a)异种移植物中,阳离子L-QD囊泡被肿瘤内治疗,提高了保留率。混合L-QD双层囊泡有望成为一种有效的递送平台,允许将广泛的治疗和诊断药物递送到癌细胞。
在另一项研究中,量子点用PEG官能化,然后装载到脂质的亲水性核心中。发现所产生的阳离子f-QD-L增强癌症细胞的细胞内化。结果表明,f-QD-L经脂质分层修饰后,可以更深入地渗透到3D多细胞球体中。此外,在实体瘤模型的瘤内注射中,f-QD-L比f-QD更有效地染色肿瘤细胞。因此,f-QD-L可以被视为一种可靠的肿瘤追踪和成像剂,无论是在体外还是体内。有趣的是,Wang等人开发了一种多功能脂质体,由脂质体载体组成,在内核中构成亲水性氧化铁,在磷脂双层中构成疏水性CdSe量子点,表面上的脂质PEG衍生物,以及附着在脂质PEG衍生物远端的cRGDyk肽。脂质-PEG衍生物的体外分析表明,其毒性低、稳定性增强、吞噬抗性提高、检测信号增强,且具有良好的肿瘤靶向性。此外,肿瘤和肝脏中多功能脂质体积聚的增加表明了它们在双模态成像中的疗效。
7.3.3 氧化铁NP
为了开发磁性NP支持的脂质双层(SLBs),羟基化阳离子肟醚脂质与核壳Fe3O4-SiO2NP(SNP)结合。以下NP负载了DOX或两亲性类似物,以研究相应阳离子SLB在MCF-7细胞中的功效。SLB-药物缀合物(50µgSNP/>5×104细胞的剂量)在暴露2小时后可在MCF-7细胞中产生极端毒性,并可容易地在癌症细胞中内化和适应,从而表明其作为磁性药物载体的能力。
7.3.4 金纳米粒子
对于siRNA的细胞内递送,Kong等人产生了阳离子脂质包覆的AuNP(L-AuNP/siRNA)。L-AuNP/siRNA的初始流体动力学直径((77.3±4.2)nm)略高于L-AuNPs((62.3±7.9)nm)。然而,与L-AuNP((59.4±4.5)mV)相比,L-AuNP/siRNA的表面电位((32.2±3.3)mV)显著降低,表明与siRNA的络合显著减轻了表面电荷。此外,原子力显微镜(AFM)高度图像证实,所得L-AuNP/siRNA偶联物具有较少的聚集球形形态。因此,L-AuNP/siRNA复合物是通过静电相互作用产生的稳定聚电解质,可以促进它们在细胞内的内化,增强基因沉默,并且比PEI(聚阳离子载体)产生的毒性更小。因此,L-AuNPs可能被用作siRNA的安全有效的细胞内载体。
7.4 与胺基改性无机纳米颗粒的相互作用
胺/酰胺的表面修饰可以催化药物在肿瘤中的成功递送。特别是无机纳米颗粒与氨基酸的功能化具有巨大的前景。在单一氨基酸中,氨基和羧基末端都可用于共轭。除了它们的生物相容性外,它们还旨在解决免疫细胞脱靶分布、形态、大小和摄取方面出现的问题。
7.4.1金纳米粒子
Biswas及其同事报道了ThomsenFriedenreich(TF)抗原通过与两种氨基酸丝氨酸和苏氨酸偶联而间接附着到AuNP上。一种名为Gal3(半乳凝素-3)的抗凋亡蛋白与TF抗原结合。在Gal3阳性的癌症中,苏氨酸修饰颗粒的细胞凋亡是丝氨酸的四倍。这可能是由于与丝氨酸相比,苏氨酸结合更高。
7.4.2 银(Ag)NP
Shi等人在组氨酸、甘氨酸和l-半胱氨酸存在下开发了柠檬酸盐包覆的AgNPs,以评估这些NPs的生物归宿和毒性。然而,甘氨酸以最低的能量结合Ag+,对转化和毒性没有明显的影响。当组氨酸以更大的结合能与Ag+结合时,AgNP的大小和Ag+的释放明显更高。Ag-组氨酸复合物的形成有助于降低AgNP的细胞毒性。此外,与Ag+结合能最高的l-半胱氨酸将AgNP完全转化为+和Ag2S沉淀,从而显著降低了AgNP的毒性。因此,颗粒尺寸和形状的减小导致它们更快地内部化,导致癌症细胞凋亡。因此,NP的大小和形态被证明会影响外源性凋亡途径的激活。
7.4.3 碳纳米管
为了减轻DOX的不良反应并增强DOX负载的CNT的抗肿瘤作用,DOX负载热敏脂质体和赖氨酸修饰的SWCNT被用作药物载体。DOX赖氨酸/SWCNTs显示出改善的药物结合效率(86.5%±3.7%)。DOX-赖氨酸/SWCNT能有效地穿透细胞膜,对人肝癌细胞(SMMC-7721)具有增强的抗肿瘤作用。使用热敏脂质体和DOX组的荷肉瘤180小鼠的肿瘤体积显著减小。用808nm的近红外激光治疗后,对SMMC-7721细胞和携带肉瘤180的小鼠的肿瘤生长抑制作用显著增强。与游离DOX相比,DOX赖氨酸/SWCNTs治疗的肉瘤180肿瘤诱导小鼠的体重、食物和水摄入量以及精神状态显著增加。因此,当与激光照射结合时,DOX赖氨酸/SWCNTs可以被用作潜在的药物递送载体。
7.4.4 硒NP
根据一项研究,用缬氨酸、赖氨酸和天冬氨酸修饰的硒(Se)NP具有更有效的抗癌作用。这些颗粒针对MCF-7、HeLa和HepG2细胞进行了测试。与丝氨酸和天冬氨酸相比,赖氨酸修饰的SeNP具有更大的抗肿瘤效力。赖氨酸修饰的硒颗粒激活了胱天蛋白酶8和胱天蛋白酶9,导致Fas和线粒体介导的细胞凋亡。ROS的增加可能是激活胱天蛋白酶的原因。值得注意的是,ROS可以由赖氨酸诱导,赖氨酸的氨基官能团是ROS的两倍。当MCF-7细胞用硒修饰的天冬氨酸NP处理时,也观察到凋亡增加。此外,Se修饰的天冬氨酸NP在癌症细胞中诱导的ROS过量产生可能是胱天蛋白酶激活和线粒体功能障碍的原因。应该指出的是,内化后,颗粒驻留在酸性溶酶体上,由于酸性pH值,氨基酸在酸性溶酶体中发生质子化,产生ROS。
7.4.5 氧化铁NP
在一项研究中,Yang及其同事用组氨酸共轭磁性聚(氨基酸)NP(H-MPNs)修饰氧化铁NP,用于DOX递送。H-MPN和MPN的TEM图像分别显示粒径在40至80nm之间。然而,根据DLS,在pH7.4时,H-MPNs的平均直径和ζ电位分别为(99.4±1.6)nm和(+13.6±1.04)mV,在pH5.2时分别为(133.7±3.7)nm和。内体中的酸性pH值诱导组氨酸电离,释放DOX。这些发现为进一步改进铺平了道路,因为只加载了6.8重量%的DOX。为了评估H-MPN的生物相容性和DOX负载的H-MPNs的治疗效果,使用HeLa细胞进行了MTT试验。将H-MPNs和MPNs以0.0625至0.25mg·mL-1的不同浓度处理至HeLa细胞,并孵育24小时。有趣的是,由于其聚(氨基酸)外壳,H-MPNs或MPNs均未表现出大量毒性作用,这表明它们具有无毒性和生物相容性。然而,与DOX负载的MPNs相比,DOX负载H-MPNs的细胞毒性增加归因于H-MPNs内体逃逸提供的DOX更大的核穿透力。因此,发现DOX负载的H-MPNs比DOX负载MPNs更有效。重要的是,在GBM中,抗血管生成治疗可能会导致急性肿瘤消退,但没有关于患者生存率改善的报道。Agemy等人设计了一种具有三种特性(CGKRK肽、(D2)肽和氧化铁成分)的纳米系统,旨在输送到肿瘤的血管系统。在小鼠中,NP的氧化铁成分允许对GBM肿瘤进行成像。在一个GBM小鼠模型中,NP的全身治疗有效地阻止了肿瘤的发展,同时显著减缓了另一个模型中的肿瘤生长。此外,观察到注射了NP的肿瘤穿透肽可以提高其治疗效果。
7.4.6 氧化钆纳米粒子(GONs)
为了确保GBM疗法的高效性(即GBM的高对比度MRI和放射敏感性),Shen等人开发了可高度运输穿过血脑屏障(BBB)的纳米材料(14nm)。这是通过用还原性牛血清白蛋白(rBSA)修饰聚丙烯酸(PAA)稳定的极小氧化钆纳米粒子(ES-GON),产生ES-GONrBSA(最初在水相中产生)来实现的。然后将RGD二聚体(RGD2,Glu-{Cyclo}2)和乳铁蛋白(LF)与ES-GON-rBSA偶联,从而产生ES-GON-rBSA-LF-RGD2复合物。为了证明血脑屏障的可移植性,通过T1加权MR图像将ES-GON-rBSA3-LF-RGD2与Magnevist进行了比较。很明显,ES-GON-rBSA3-LF-RGD2在体内穿过血脑屏障,可以获得脑增强MRI对比,但在这种Gd浓度下,Magnevist几乎不会在脑中积聚。此外,使用T1加权MRI在体内研究了ES-GON-rBSA3-LF-RGD2对胶质母细胞瘤的疗效。在携带U-87MG肿瘤的裸鼠中,静脉注射ES-GON-rBSA3-LF-RGD2,然后在注射后的不同时间点进行T1加权MR成像。静脉注射前,肿瘤的MRI信号非常微弱,但静脉注射后有所改善,在注射ES-GON-rBSA3-LF-RGD2后12小时达到峰值。ES-GON-rBSA-LF-RGD2具有高度的生物相容性,由于其体积小(13.4nm)以及LFDD受体介导的转胞作用,可以穿过小鼠的血脑屏障。原位GBM研究的结果证实,ES-GON-与三苯基鏻(TPP)抗生素结合肽(KLAKLAK)2(TPep)用于产生所需的MSNrBSA3-LF-RGD2可以在GBM中积聚,并作为放射增敏剂促进放射治疗。
7.4.7
Taratula等人通过PEG间隔物结合促黄体生成素释放激素(LHRH,一种释放促性腺激素的激素十肽),结合抗癌药物(DOX和顺铂),用两种形式的siRNA作为泵和非泵细胞耐药性的抑制剂,从而修饰了MSN表面。原位小鼠模型用于测试基于MSN的DDS实现活性成分局部吸入肺递送的能力。吸入基于MSN的DDS的局部给药导致纳米载体在肺部的优先积聚(73%),是静脉注射(5%)的14.6倍。因此,MSN被阻止进入体循环并在其他组织中积聚。总体而言,静脉注射(7%)后,基于MSN的每个器官的DDS在肝脏(73%)、肾脏(15%)和脾脏(15%)中最为丰富。相比之下,单次吸入后,肝脏、肾脏和脾脏中基于MSN的DDS的百分比分别为17%、9%和1%。
值得注意的是,当与肽基序结合时,NP可以具有改善的药代动力学特征。例如,Fei等人报道了构建Arg-Gly-Asp(RGD)偶联的脂质体-中空二氧化硅杂化物(RGD-LP-CHMSN)来递送和释放三氧化二砷(ATO),其肿瘤靶向作用受到剂量限制毒性和药代动力学的阻碍。在MCF-7、HepG2和LO2细胞中,RGD-LP-CHMSN 表现出最小的毒性和优异的生物相容性。RGD-LP-CHMSN-ATO显示细胞摄取增加。此外,该杂化物显示出半最大抑制浓度(IC50)的降低。根据药代动力学分析,与ATO组相比,RGD-LP-CHMSN-ATO组的半衰期增加了1.7倍,曲线下面积增加了2.4倍。此外,这些纳米载体增强了ATO在H22肿瘤异种移植物模型中的靶向潜力。目前,具有官能团的(三烷氧基)硅烷通常用于修饰MSN表面,以附着各种分子,包括siRNA、肽的靶配体和聚合物。例如,用(3-巯基丙基)三甲氧基硅烷修饰的MSN,用2,2-二吡啶基二硫化物处理的MSN和癌症,如前几节所示。化疗药物可以通过无机NP递送,从而实现更有效的药物或基因递送,同时超越化疗的限制。例如,主动和被动靶向增强了载药纳米载体在癌症细胞中的积累,而不是在健康组织中。通过不同的策略和相互作用修饰无机纳米载体以装载治疗剂和显像剂,提高了其对抗癌症细胞的有效性。总结了几种负载药物的无机纳米载体对游离药物的疗效。
TPep。盐酸溶液中的拓扑替康(TPT,一种拓扑异构酶抑制剂)可以通过在室温下搅拌过夜而掺入PBS中的MSN-TPep中,以对抗卵巢癌症、癌症和其他癌症。SEM和TEM拍摄的显微照片显示,研究中使用的MSN的平均粒径为120nm,具有单分散分布。然而,DLS数据显示粒径稍大,粒径分布窄。对于肿瘤酸敏感的聚乙二醇化阴离子聚合物,带正电荷的MSNTPepNP表现出快速进入细胞(由于它们对带负电荷的细胞膜的吸引力),从血液中快速消除,并增强肿瘤中的积聚。随后,将NP锚定在聚乙二醇封端的2,3-二甲基马来酸酐改性的聚L-赖氨酸(PEG-PLL(DMA))上,通过静电相互作用产生MSNTPep/PEG-PLL(DMA)NP。所制备的MSNTPep/PEGPLL(DMA)NP的表面电位为212.4mV。此外,MSNTPep/PCEPLL(DMA)NPs在体外显示出肿瘤细胞内化增加、线粒体损伤能力增强和优异的抗肿瘤效率。此外,与TPT结合可显著抑制肿瘤生长。
8.载药无机纳米载体对游离药物的疗效
无机NP在治疗制剂开发方面非常有效。例如,具有−NH2基团的药物分子可能会与−COOH基团结合产生巨大的聚集体。此外,离子相互作用会受到pH值的影响,使其在酸性环境中不稳定。
9.结论和未来展望
在应对与无机NP相关的挑战方面取得的进展(包括脱靶药物分布、连续生物屏障、血液中的不稳定性)利用了它们在癌症治疗中的应用。使用无机NP递送治疗性货物,从蛋白质到小分子、肽和核酸,极大地改善了新兴疗法的翻译。相反,用于开发和优化基于NP的治疗平台的不同相互作用或策略(物理包埋、离子相互作用、共价键、基于亲和力的相互作用和插层)已被调整,以提高其在新的和现有的药物递送系统中的疗效。基于这些相互作用和方法,基于无机NP的治疗平台可以具有各种可修改的特征(包括表面特性、尺寸、形态和反应性),可以量身定制以优化其靶向递送的性能。通过各种相互作用用不同的偶联物修饰无机NP,开发不同的药物递送系统的先入为主的想法仍在进行许多研究。与其他相互作用相比,亲和力(生物素-亲和素)相互作用具有极高的亲和力,因为它由几个亲水性氢键组成。重要的是,离子装载药物的相互作用可能会给肿瘤微环境。尽管存在这些缺点,但涉及用胺/酰胺基团、阳离子大分子(聚合物、脂质)或钙掺杂的CaP、CaCO3颗粒对NP进行表面改性的离子相互作用为靶向药物递送提供了动力。通过共沉淀或阴离子交换反应(如LDH)将治疗剂掺入多孔NP或插入基于NP的平台可以改善基因和药物递送。与物理封装相反,共价键具有明显的优势,包括提高治疗效果、增强药物在体内的驻留、改善生物分布、减少释放时间和减轻全身毒性的能力。因此,NP的共价官能化在病变部位递送和释放有效载荷方面起着至关重要的作用。尽管基于阳离子聚合物或基于脂质的NP上的表面电荷可能会增强细胞对它们的摄取,但这些药物递送系统往往会聚集、失去稳定性并变得容易产生毒性,并被MPS消除。据信,需要大剂量的载药颗粒来提供治疗剂量是MPS器官中带正电NP分布的主要决定因素,导致阳离子NP聚集。确实,阳离子聚合物或基于脂质的NP本身不会造成任何伤害,但它们在聚集时能够在肺部引起栓塞。带负电荷的生物膜和阳离子聚合物或基于脂质的NP之间的优越相互作用可能被证明是不方便的,因为颗粒和红细胞膜表现出相同的明显亲和力。值得注意的是,一个相关的事件是循环中阳离子颗粒的调理和巨噬细胞吸收加速。为了确保循环的延长,颗粒必须尽可能微小和中性。
为了有效地将治疗和成像分子输送到作用部位,需要更多的体外研究和解释,以及准确的非侵入性监测(随着时间的推移)来评估NP平台的疗效(通过这些方法或相互作用制备)。因此,有必要基于这些方法或相互作用对NP设计进行更深入的分析,并对NP在体内的命运进行更详细的调查,以提高这些声明的特异性。此外,小动物模型通常是临床研究的主要临床前模型,这进一步使这个问题复杂化,因为它们不能准确地代表人类。因此,在癌症治疗的临床试验开发阶段,必须解决和克服这些限制,评估安全性,筛选治疗效果,并实施严格的控制。
表1药物与载药无机纳米载体疗效比较

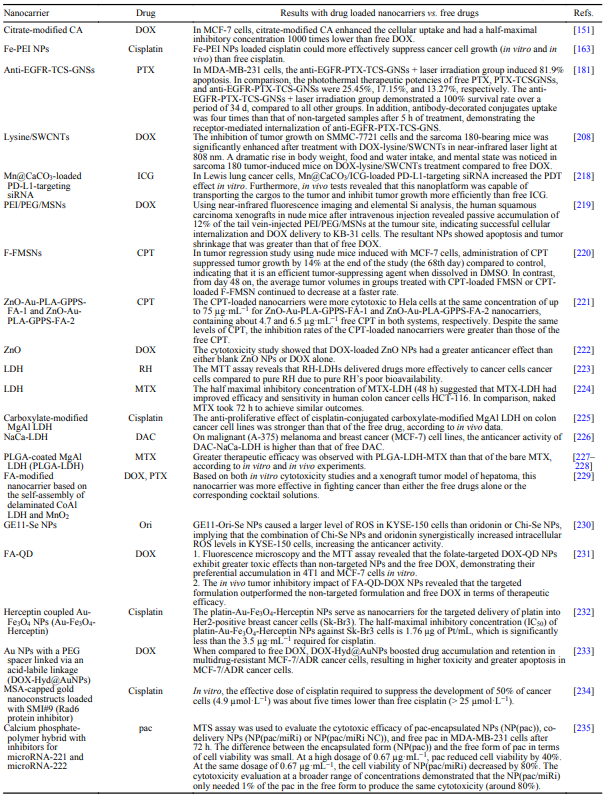
![]()
正如本综述所强调的那样,不同策略或相互作用(如物理包埋到多孔/中空纳米结构中、与天然和表面改性的NP的离子相互作用、共价键合、基于亲和力的相互作用以及通过共沉淀或阴离子交换反应的插层)对改性/未改性无机NP中的负载疗法的前所未有的影响显示出了优于其他传统NP药物递送系统的明显优势。此外,这些方法导致的物理化学特性的变化是改善药代动力学参数的支柱(如减少不加选择的组织分布、延长血液停留时间、降低与药物相关的毒性等),这阐明了针对特定部位递送的创新药物递送平台的设计。尽管大多数先前存在的基于NP的药物递送平台在临床转化中都不成功,但在不久的将来仍有希望建立新的有前景的治疗癌症的平台。
原文链接:https://link.springer.com/article/10.1007/s11706-022-0604-x